
mar
25
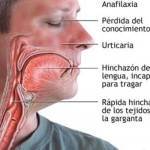 Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
El trabajo, publicado en la revista científica Frontiers in Immunology, destaca la identificación de varios casos portadores de la misma variante genética (T328K gen F12), todos ellos concebidos mediante reproducción asistida a través de un único donante.
Angioedema hereditario
El angioedema hereditario es una enfermedad genética muy poco frecuente que provoca hinchazón en la piel, aparato digestivo o vías respiratorias; unos síntomas que afectan al 60-80 % de las mujeres portadoras, frente a una probabilidad muy baja en los hombres.
La investigación arrancó con una paciente que presentaba episodios recurrentes de hinchazón en la cara, sin responder a los tratamientos antialérgicos habituales, y cuyo análisis genético identificó la variante T328K.
Esta fue también detectada posteriormente en otras personas concebidas con el esperma del mismo donante, mientras que no estaba presente en la línea materna analizada.
Ante la sospecha de que la mutación genética podía proceder del padre, los investigadores contactaron con la clínica de fertilidad y, tras coordinar un estudio con el banco de esperma, pudieron confirmar que este era portador heterocigoto de la variante.
Notificación de la clínica
La clínica notificó entonces a las mujeres que habían utilizado el esperma de dicho donante, un procedimiento que permitió identificar a más portadores, algunos de ellos sin manifestaciones sintomáticas.
«Desde el punto de vista genético, este caso es especialmente relevante porque demuestra cómo una variante patogénica puede pasar desapercibida en un donante asintomático y transmitirse a varios descendientes», explica el jefe del grupo de Inmunología Traslacional del Vall d’Hebron Institut de Recerca (VHIR), Roger Colobran.
Los investigadores subrayan que este caso pone de relieve los retos que plantean las enfermedades genéticas hereditarias asintomáticas en los programas de donación de gametos, ya que el cribado genético de los donantes se centra principalmente en enfermedades infecciosas.
En este sentido, señalan que, en regiones donde esta variante es más prevalente, podría valorarse el cribado dirigido al gen F12.
20 marzo 2026 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
dic
22
 Ante el aumento de circulación del virus de influenza A(H3N2) subclado K (J.2.4.1) en varias regiones del mundo, la Organización Panamericana de la Salud (OPS) llamó a los países de las Américas a fortalecer la vigilancia y la vacunación.
Ante el aumento de circulación del virus de influenza A(H3N2) subclado K (J.2.4.1) en varias regiones del mundo, la Organización Panamericana de la Salud (OPS) llamó a los países de las Américas a fortalecer la vigilancia y la vacunación.
La OPS también instó a tratar oportunamente los casos y a preparar los servicios de salud ante una posible actividad temprana o más intensa de enfermedades respiratorias.
Datos recientes muestran que la circulación del subclado K ha crecido rápidamente en Europa y en varias naciones de Asia, donde representa una proporción importante de los virus de influenza A(H3N2) analizados.
Las autoridades sanitarias de esos países no han reportado cambios significativos en la gravedad clínica, pero históricamente las temporadas dominadas por A(H3N2) suelen asociarse con mayor impacto en personas adultas mayores.
En América del Norte, Estados Unidos y Canadá registran también un aumento progresivo de detecciones del subclado K.
Hasta la fecha no se ha observado una circulación similar en América del Sur, según los datos disponibles en la plataforma de la Iniciativa Global para Compartir todos los Datos de la Gripe.
La OPS señaló que la evolución genética observada en el subclado K forma parte del proceso natural de variación del virus de la influenza estacional.
Aunque la evidencia sobre la efectividad de la vacuna de esta temporada aún es limitada, informes preliminares de Europa indican que la vacunación ofrece una protección parecida a los años anteriores frente a la enfermedad grave, incluida la hospitalización.
En este contexto, la OPS instó a los Estados Miembros a mantener y fortalecer la vigilancia epidemiológica, virológica y genómica, garantizar una alta cobertura de vacunación, tratar oportunamente a los casos y reforzar la preparación de los servicios de salud ante la posibilidad de una actividad temprana o más intensa durante la temporada 2025-2026 en el hemisferio norte.
12 diciembre 2025 | Fuente: Prensa Latina | Tomado de | Noticia
dic
22
 A propósito de la actual expansión mundial de la variante de la influenza A H3N2 subclado K, expertos señalan el dinamismo de su circulación entre hemisferios en temporada de gripe.
A propósito de la actual expansión mundial de la variante de la influenza A H3N2 subclado K, expertos señalan el dinamismo de su circulación entre hemisferios en temporada de gripe.
Según afirma Marcus Pereira, médico especialista en enfermedades infecciosas del Centro Médico Irving de la Universidad de Columbia, en Nueva York, citado por la publicación National Geographic, existe un efecto ping-pong entre las temporadas de gripe del hemisferio norte y del sur.
El galeno asevera que «lo que hay allí acaba llegando hasta nosotros, y viceversa».
Se refiere el especialista a la vuelta al mundo que registra la denominada versión subclado K, la cual ha sufrido cambios genéticos significativos que facilitan el contagio entre las personas y, por lo tanto, su propagación.
Por su parte la médica epidemióloga Danuta Skowronski, investigadora de la gripe en el Centro para el Control de Enfermedades de Columbia Británica, plantea que «debido precisamente a estos cambios genéticos, «nuestros anticuerpos no la detectarán tan fácilmente», citó la fuente.
Precisó National Geographic que Skowronski fue la primera experta en señalar las mutaciones en la revista Journal of the Association of Medical Microbiology and Infectious Disease Canada, en octubre.
En su criterio, la situación es compleja porque estos cambios genéticos se produjeron demasiado tarde para tenerlos en cuenta en las formulaciones de las vacunas contra la gripe.
Los especialistas prevén que la gripe predomine en Estados Unidos y Canadá durante estas vacaciones de invierno.
Apuntan que ahora es aún más importante vacunarse contra la gripe, ya que el virus, más contagioso, infectará a un mayor número de personas, y aumentará el número de los que enfermarán gravemente.
Según la publicación, en el hemisferio sur las personas que se vacunaron contra la gripe este verano tuvieron casi un 50 % menos de hospitalizaciones, de acuerdo con análisis de los Centros para el Control y la Prevención de Enfermedades de Estados Unidos (CDC).
«Pero no se sabrá si este país y Canadá tendrán los mismos resultados, o peores, hasta al menos mediados de enero, cuando más personas habrán estado expuestas y la mayoría de las que quieren vacunarse lo habrán hecho», dijo Skowronski al respecto.
Las cifras publicadas indican que las tasas de gripe ya han comenzado a aumentar en Estados Unidos.
Pero, aunque la vacuna de este año no se ajusta perfectamente a la cepa dominante, los expertos insisten en que sigue siendo importante vacunarse, así como tomar otras medidas para combatir una avalancha gripal, apuntó National Geographic.
12 diciembre 2025 | Fuente: Prensa Latina | Tomado de | Noticia
dic
10
 Expertos y autoridades sanitarias enfatizan hoy en la importancia de la vacunación ante la irrupción de la nueva variante de gripe H3N2 subclado K en Europa.
Expertos y autoridades sanitarias enfatizan hoy en la importancia de la vacunación ante la irrupción de la nueva variante de gripe H3N2 subclado K en Europa.
Investigaciones sobre la incidencia actual de la infección indican que el nuevo subtipo es mucho más fácil de transmitir que las cepas de gripe conocidas hasta ahora, reportó Euronews.
La expansión de esta variante, altamente transmisible, adelantó la temporada en el continente.
El Centro Europeo Para el Control y la Prevención de Enfermedades (ECDC por sus siglas en inglés) continúa alertando sobre la intensidad de la nueva mutación y la protección de la inmunización.
Sobre todo, insiste en la importancia de vacunarse para las personas de alto riesgo a fin de evitar llegar a la gravedad.
Se prevé en la región un fuerte aumento de casos y los especialistas advierten del riesgo de infecciones dobles (gripe y gripe aviar), informa Euronews.
De acuerdo con medios europeos, los síntomas de esta variante gripal suelen ser similares a cualquier otra A(H3N2).
Se puede experimentar fiebre superior a 38ºC, escalofríos, tos y dolor muscular, entre otros síntomas.
Otros países del hemisferio norte donde esta mutación es la predominante son Japón, Reino Unido, Estados Unidos y Canadá.
En Europa, el ECDC prevé que un número de infecciones superior al habitual añadiría más presión a los sistemas sanitarios de los países del bloque.
01 diciembre 2025 | Fuente: Prensa Latina | Tomado de | Noticia
abr
22
 Este descubrimiento ofrece «respuestas» y «esperanza» a miles de familias que llevan años buscando el diagnóstico de una serie de trastornos de base genética que se manifiestan en la primera infancia, y que pueden provocar situaciones altamente discapacitantes.
Este descubrimiento ofrece «respuestas» y «esperanza» a miles de familias que llevan años buscando el diagnóstico de una serie de trastornos de base genética que se manifiestan en la primera infancia, y que pueden provocar situaciones altamente discapacitantes.
«Sabemos por años de experiencia en el apoyo a pacientes y familias con enfermedades genéticas raras que recibir un diagnóstico como este puede cambiar la vida y ser el primer paso para poner en marcha el apoyo y la atención adecuados», ha destacado el coordinador de la infraestructura IMPaCT-Genómica, Ángel Carracedo, que ha participado en la investigación junto con los programas de enfermedades raras no diagnosticadas ENoD-CIBERER y URDcat.
Tras ello, ha resaltado que la red de hospitales participantes en la infraestructura IMPaCT-Genómica y otros programas de enfermedades no diagnosticadas pueden aprovecharse de estos «punteros» descubrimientos y mejorar «rápidamente» las tasas de diagnóstico a nivel local.
Los científicos han señalado que los avances en las técnicas de secuenciación del genoma completo y la exploración de regiones no codificantes en grandes cohortes de pacientes han supuesto reciente y gran avance en la genética de los trastornos del neurodesarrollo.
«Trabajar con datos genómicos de cohortes grandes de pacientes bien caracterizados fenotípicamente, con sistemas de análisis masivos y de intercambio de datos, brinda la oportunidad de acelerar el descubrimiento de nuevos hallazgos y validarlos rápidamente, como se demuestra en este estudio en el que colaboramos», ha señalado Carracedo.
El equipo de investigadores, a partir del descubrimiento del síndrome RNU4-2/ReNU el año pasado, uno de los tipos monogénicos más comunes de los trastornos del neurodesarrollo, ha logrado identificar este nuevo síndrome relacionado que está causado por mutaciones en el gen no codificante RNU2-2.
Tras analizar los datos de miles de personas con trastornos del neurodesarrollo se han podido hallar nueve casos con mutaciones de novo en este gen, a los que se han sumado otros 16 casos fruto del análisis masivo de otras ocho colecciones de enfermedades raras no diagnosticadas.
La identificación de las mutaciones en RNU2-2 como nueva causa de los trastornos del neurodesarrollo es «especialmente notable» debido a que consolida la importancia biológica de estos genes en ese tipo de afecciones, pues tienden a producirse espontáneamente en lugar de heredarse de los progenitores.
Por su parte, la colaboradora de la coordinación de ENoD-CIBERER e IMPaCT-Genómica, Beatriz Morte, ha expresado que este descubrimiento también posibilita nuevas investigaciones encaminadas a explorar los mecanismos moleculares subyacentes al trastorno.
La experta también ha explicado que los síndromes RNU4-2/ReNU y RNU2-2 comparten similitudes, pero que los pacientes con síndrome RNU2-2 suelen estar más afectados por la epilepsia.
«Se estima que la prevalencia del trastorno RNU2-2 es aproximadamente el 20 % de la del síndrome RNU4-2/ReNU, uno de los tipos monogénicos más comunes de los trastornos del neurodesarrollo. Esto significa que debe haber miles de familias afectadas en todo el mundo», ha agregado.
Esta investigación ha servido igualmente para identificar una mutación distinta en RNU2-2 que tiende a aparecer en individuos no afectados a medida que envejecen, lo que puede tener implicaciones para las afecciones relacionadas con la edad.
Los programas españoles involucrados en el estudio son IMPaCT-Genómica, con cerca de 2 000 pacientes que provienen de todas las comunidades autónomas, y que es una iniciativa del Instituto de Salud Carlos III gestionada por el Centro de Investigación Biomédica en Red (CIBER), el Programa de Enfermedades no Diagnosticadas del CIBER de Enfermedades Raras (ENoD-CIBERER) y el programa de enfermedades no diagnosticadas de la Generalitat de Catalunya (URDCat). El estudio también ha contado con la colaboración de equipos de Reino Unido, Bélgica, Países Bajos e Islandia.
15 abril 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jul
19
 Investigadores andaluces han identificado nuevas variantes de un gen denominado COQ7, relacionado con la generación de enfermedades neurodegerativas llamadas mitocondriales, lo que abre una nueva vía al tratamiento de estas patologías en niños que la sufren.
Investigadores andaluces han identificado nuevas variantes de un gen denominado COQ7, relacionado con la generación de enfermedades neurodegerativas llamadas mitocondriales, lo que abre una nueva vía al tratamiento de estas patologías en niños que la sufren.
Se trata de un estudio del Centro Andaluz de Biología del Desarrollo (CABD) dirigido por el catedrático de Biología Celular de la Universidad Pablo de Olavide Carlos Santos Ocaña, que ha identificado nuevas variantes genéticas del COQ7 y su relación con la deficiencia de Coenzima Q10 en pacientes pediátricos, lo que hará que se puedan mejorar el diagnóstico y tratamiento de enfermedades mitocondriales.
En un comunicado, la Universidad Pablo de Olavide (UPO) ha recordado que las mitocondriales, que afectan principalmente la producción de energía en las células, son un grupo diverso de enfermedades raras, poco frecuentes, que pueden causar una amplia gama de síntomas y afectar varios órganos del cuerpo, y se estima que existen alrededor de 300 diferentes, cada una de ellas con sus particularidades.
En el estudio, los pacientes que portan variantes (mutaciones) del gen COQ7 padecen una enfermedad mitocondrial producida por una deficiencia de COQ10, patología que se caracteriza por producir una serie de alteraciones moleculares, comunes a todos los pacientes, que son propias de una grave disfunción mitocondrial.
Sin embargo, cada uno de los pacientes muestran una diferente combinación de variantes génicas que son responsables de alteraciones fisiológicas específicas que explican los diversos fenotipos de la enfermedad y, por tanto, de la gravedad de esta.
Debido a su complejidad y la variabilidad de sus síntomas, suponen un gran desafío tanto para la comunidad médica y científica como para los enfermos y sus familias, por lo que su investigación es crucial para avanzar en su conocimiento y desarrollar mejores diagnósticos y tratamientos.
En esta línea, Ocaña lidera un estudio que abre la vía a identificar marcadores para el diagnóstico, la gravedad y pronóstico de la enfermedad, posibilitando además cierta personalización en terapias aplicables a cada paciente.
El trabajo cierra el diagnóstico, clínico y molecular, de tres pacientes pediátricos que mostraban una enfermedad mitocondrial: en los tres casos la patología se produce por la presencia de variantes patológicas del gen COQ7, que codifica una proteína que participa de la síntesis de Coenzima Q10.
Esta molécula es esencial para el correcto funcionamiento de las células y la salud en general; su papel en la producción de energía, la protección antioxidante, y el apoyo a la salud cardiovascular y muscular la hace crucial para el bienestar humano.
Además de cerrar el diagnóstico con pruebas funcionales, el equipo de investigación realizó estudios adicionales para intentar explicar la heterogeneidad de los síntomas mostrados por los pacientes, a pesar de compartir variantes del mismo gen y de mostrar de forma común una deficiencia de CoQ10.
Como explica el investigador Carlos Santos, se ha diagnosticado definitivamente a tres pacientes que mostraban una enfermedad mitocondrial con deficiencia de Coenzima Q10 originada por diferentes variantes (mutaciones) del gen COQ7, y aunque los tres presentan dicha deficiencia, «realmente sus síntomas son muy heterogéneos, al igual que la gravedad de la enfermedad», que va desde un caso muy grave con fallecimiento temprano del paciente «a un caso mucho más leve que mejoró con un tratamiento con CoQ10″.
El estudio ha permitido demostrar así que los cambios estructurales generados por cada variante identificada en los pacientes alteran la función de la proteína COQ7 de manera específica, y que la combinación de estas variantes, lo que determina el contexto genético del paciente, explica no solo la gravedad de la enfermedad, sino también el efecto tan variable que muestra la terapia disponible para el tratamiento de la deficiencia de CoQ10.
La investigación, publicada en Journal of Inherited Metabolic Disease, ha contado con la participación de los hospitales Sant Joan de Déu, Vall d’Hebron, Santiago de Compostela y La Fe de Valencia, y de varios equipos del Centro de Investigación Biomédica en Red de Enfermedades Raras (Ciberer), coordinados desde el grupo del CABD ‘Regulación de la síntesis de coenzima Q y sus implicaciones en la salud mitocondrial’.
17 julio 2024|Fuente: EFE|Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2024. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
