
abr
21
 La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
De acuerdo con la literatura médica, es considerada poco frecuente y que aqueja a hombres y mujeres de cualquier edad, sin importar el origen étnico, e incluso puede originarse en cualquier etapa de vida del paciente desde lactantes hasta adultos.
Es una enfermedad neuromuscular hereditaria autosómica recesiva y de origen genético, causada por un gen que genera el déficit de la enzima alfa-glucosidasa ácida o maltasa ácida.
Ello permite la acumulación de una sustancia en los músculos denominada glucógeno, ocasionando pérdida de masa muscular, así como debilidad muscular en las extremidades inferiores.
Igualmente puede afectar los músculos del tronco, brazos y hombros, y la gravedad y síntomas varían en cada paciente.
Entre los malestares relacionados con esta dolencia genética están la dificultad para levantarse, al estar sentado, para caminar, subir escaleras, masticar y la debilidad muscular progresiva.
Una persona aquejada por la Enfermedad de Pompe, también puede presentar reflujo gastroesofágico, disnea, caídas frecuentes, agitación tras realizar esfuerzo físico, dolor de cabeza por las mañanas y somnolencia durante el día debido a la ortopnea.
El nombre de esta dolencia es en honor al patólogo holandés Joannes Pompe, quien presentó el primer caso en el año 1930, y cada 15 de abril es celebrado el Día Internacional de la Enfermedad de Pompe.
15 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
mar
25
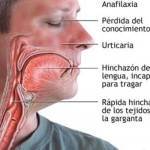 Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
El trabajo, publicado en la revista científica Frontiers in Immunology, destaca la identificación de varios casos portadores de la misma variante genética (T328K gen F12), todos ellos concebidos mediante reproducción asistida a través de un único donante.
Angioedema hereditario
El angioedema hereditario es una enfermedad genética muy poco frecuente que provoca hinchazón en la piel, aparato digestivo o vías respiratorias; unos síntomas que afectan al 60-80 % de las mujeres portadoras, frente a una probabilidad muy baja en los hombres.
La investigación arrancó con una paciente que presentaba episodios recurrentes de hinchazón en la cara, sin responder a los tratamientos antialérgicos habituales, y cuyo análisis genético identificó la variante T328K.
Esta fue también detectada posteriormente en otras personas concebidas con el esperma del mismo donante, mientras que no estaba presente en la línea materna analizada.
Ante la sospecha de que la mutación genética podía proceder del padre, los investigadores contactaron con la clínica de fertilidad y, tras coordinar un estudio con el banco de esperma, pudieron confirmar que este era portador heterocigoto de la variante.
Notificación de la clínica
La clínica notificó entonces a las mujeres que habían utilizado el esperma de dicho donante, un procedimiento que permitió identificar a más portadores, algunos de ellos sin manifestaciones sintomáticas.
«Desde el punto de vista genético, este caso es especialmente relevante porque demuestra cómo una variante patogénica puede pasar desapercibida en un donante asintomático y transmitirse a varios descendientes», explica el jefe del grupo de Inmunología Traslacional del Vall d’Hebron Institut de Recerca (VHIR), Roger Colobran.
Los investigadores subrayan que este caso pone de relieve los retos que plantean las enfermedades genéticas hereditarias asintomáticas en los programas de donación de gametos, ya que el cribado genético de los donantes se centra principalmente en enfermedades infecciosas.
En este sentido, señalan que, en regiones donde esta variante es más prevalente, podría valorarse el cribado dirigido al gen F12.
20 marzo 2026 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
ene
26
 La Universidad Séchenov de Moscú informó hoy que un grupo de especialistas del centro crearon la primera tecnología mundial de biocamuflaje de vectores virales. Según la comunicación la tecnología permite la administración repetida y segura de medicamentos de terapia génica sin riesgo de neutralización por el sistema inmunológico.
La Universidad Séchenov de Moscú informó hoy que un grupo de especialistas del centro crearon la primera tecnología mundial de biocamuflaje de vectores virales. Según la comunicación la tecnología permite la administración repetida y segura de medicamentos de terapia génica sin riesgo de neutralización por el sistema inmunológico.
«Los científicos de la Universidad Séchenov desarrollaron una tecnología única de biocamuflaje de vectores virales que, por primera vez en el mundo, permite la administración repetida de fármacos de terapia génica sin el riesgo de ser neutralizados por el sistema inmunológico», reza la nota.
Según la universidad, esta tecnología ofrece una solución fundamental a uno de los principales problemas de la terapia génica moderna, la imposibilidad de readministrar los fármacos.
Igualmente aclara que, pese a los éxitos de la terapia génica en el tratamiento de enfermedades hereditarias graves, como la atrofia muscular espinal, enfermedades de almacenamiento y mitocondriales, todos los medicamentos existentes basados en vectores virales solo pueden aplicarse una vez.
Esto se debe a que el sistema inmunológico «recuerda» el virus lo neutraliza de inmediato en aplicaciones posteriores, reduciendo drásticamente la eficacia del tratamiento.
Según el comunicado, la tecnología, basada en el uso de membranas celulares, hace que el vector viral permanezca «invisible» para los anticuerpos, sin perder su capacidad de transportar genes terapéuticos al órgano objetivo, donde es necesario restaurar la función del gen defectuoso.
La tecnología es aplicable a la mayoría de los vectores virales utilizados en la terapia génica moderna.
De acuerdo con los investigadores, este avance abre el camino a una terapia génica completa y múltiple, lo cual es especialmente importante para enfermedades que requieren tratamiento prolongado o de por vida.
Además, se destaca que el biocamuflaje reducirá significativamente el costo del tratamiento al eliminar la necesidad de desarrollar nuevos vectores para cada readministración.
20 enero 2026 | Fuente: Prensa Latina | Tomado de | Noticia
ene
6
 Científicos del Imperial College London desarrollaron una herramienta de Inteligencia Artificial (IA) para identificar genes vinculados a enfermedades y acelerar la búsqueda de fármacos para patologías cardíacas.
Científicos del Imperial College London desarrollaron una herramienta de Inteligencia Artificial (IA) para identificar genes vinculados a enfermedades y acelerar la búsqueda de fármacos para patologías cardíacas.
Según los científicos, esta tecnología podría a futuro contribuir al logro de una atención más personalizada y tratamientos mejor ajustados al corazón de cada paciente, reportó Euronews.
El equipo combinó escáneres detallados del corazón con grandes bases de datos médicas.
La herramienta, llamada CardioKG, se construyó con datos de imagen cardíaca de miles de personas del Biobanco del Reino Unido.
Fueron analizados pacientes con afecciones como fibrilación auricular, insuficiencia cardíaca e infartos, además de voluntarios sanos.
Khaled Rjoob, primer autor del estudio e investigador en ciencia de datos en el Imperial College London, comentó que, a partir de este trabajo, amplían el grafo de conocimiento hasta convertirlo en un marco dinámico, centrado en el paciente, que capte trayectorias reales de la enfermedad, citó Euronews.
El experto dijo que ellos abrirán más posibilidades para el tratamiento personalizado y para prever cuándo es probable que se desarrollen las enfermedades.
Concretamente los investigadores dicen que pueden hacer predicciones más precisas sobre qué medicamentos podrían ayudar a personas con patologías cardíacas concretas.
03 enero 2025 | Fuente: Prensa Latina | Tomado de | Noticia
dic
22
 El ADN basura es el nombre que se le da a partes menos conocidas del genoma humano. Por mucho tiempo, los científicos pensaron que esas largas secuencias no hacían nada útil porque no sirven para fabricar proteínas.
El ADN basura es el nombre que se le da a partes menos conocidas del genoma humano. Por mucho tiempo, los científicos pensaron que esas largas secuencias no hacían nada útil porque no sirven para fabricar proteínas.
En la actualidad, ya se sabe que ese ADN sí puede tener funciones muy importantes, como contener instrucciones que ayudan a controlar lo que sucede dentro de las células.
Un grupo de la Universidad de Nueva Gales del Sur y la Universidad Monash en Australia descubrió que en el ADN basura hay interruptores especiales llamados potenciadores. Los resultados se publicaron en la revista Nature Neuroscience y podrían cambiar la lucha contra el Alzheimer, según los autores.
Esos interruptores controlan cómo trabajan células del cerebro llamadas astroglías, que ayudan a mantener las neuronas sanas.
Nicole Green, la principal autora del estudio, explicó que su equipo “usó una técnica llamada CRISPRi para apagar estos potenciadores en las células y ver si cambiaba el comportamiento de ciertos genes”.
El Alzheimer es la causa más común de demencia en personas mayores. Afecta la memoria, el pensamiento y, con el tiempo, la capacidad de realizar tareas cotidianas.
Según la Organización Mundial de la Salud, más de 55 millones de personas en el mundo tienen demencia, y entre un 60 % y un 70 % de esos casos se deben a la enfermedad de Alzheimer.
Este trastorno representa un desafío gigantesco no solo para los pacientes, sino también para sus familias y los sistemas de salud.
Los investigadores de Australia analizaron cerca de 1 000 secuencias de ADN catalogadas como potenciadores, que son como interruptores a distancia.
Green contó que “si al apagar uno de estos potenciadores veíamos que el funcionamiento de un gen cambiaba, sabíamos que era importante y podíamos descubrir qué gen estaba afectando. Eso ocurrió en unos 150 de los 1 000 que probaron”.
Lo sorprendente, resaltó, “es que muchos de estos potenciadores están involucrados en genes relacionados con el Alzheimer”.
Este hallazgo facilita la búsqueda de las partes del ADN que podrían influir en el Alzheimer. Antes, había miles de lugares posibles para investigar, pero ahora pueden enfocarse en los 150 interruptores más importantes.
Green comentó que “pasar de tener 1 000 posibles interruptores a solo 150 ayuda mucho, porque así los científicos ya saben mejor en dónde buscar pistas sobre la enfermedad”.
En tanto, Irina Voineagu, supervisora del estudio, explicó que este listado de potenciadores sirve también para entender otras enfermedades.
Dijo que, al buscar respuestas sobre problemas como la hipertensión, la diabetes o el Alzheimer, muchas veces las partes del ADN más importantes no están dentro de los genes, sino entre ellos, en estos interruptores especiales.
El equipo combinó CRISPRi con secuenciación de ARN de célula única, lo que les permitió revisar la función de casi 1 000 potenciadores a la vez, algo que nunca se había hecho antes en células cerebrales.
Además, los datos ya están ayudando a entrenar programas informáticos que predicen cómo funciona el ADN.
“Este conjunto de datos puede ayudar a las computadoras a mejorar sus predicciones sobre la función de los potenciadores”, contó Voineagu.
Poder manipular estos interruptores da esperanza para crear terapias genéticas más precisas y seguras, siempre de acuerdo con estos científicos.
Si bien todavía faltan años para que esto llegue a los hospitales, Voineagu destacó que el primer medicamento de edición genética aprobado para una enfermedad de la sangre, la anemia falciforme, ya actúa sobre un potenciador específico.
Green concluyó que investigar estos interruptores puede ser clave para la medicina personalizada del futuro: “Queremos descubrir qué potenciadores podemos usar para prender o apagar genes en solo un tipo de célula cerebral y hacerlo de manera muy controlada”, afirmó.
18 diciembre 2025 | Fuente: Infobae | Tomado de | Noticia
oct
8
 De este modo, la Sociedad Balear de Laboratorio Clínico, Asociación Canaria del Laboratorio Clínico (ACLAC), Asociación de Analistas Clínicos de Extremadura (ANCLEX), Asociación Valenciana de Especialistas en Laboratorio Clínico (AVELAC), Sociedad Castellanomanchega de Medicina de Laboratorio (LABCAM), Sociedad Asturiana de Bioquímica y Análisis Clínicos – Medicina de Laboratorio (SAByAC ML), Sociedad Andaluza de Análisis Clínicos, Medicina del Laboratorio (SANAC) y Sociedad Castellanoleonesa de Análisis Clínicos (SCLAC) y la Sociedad de Medicina de Laboratorio de la región de Murcia (SOMELMUR), apuestan por la creación de un Diploma de Acreditación Avanzada para esta área funcional que consideran «constituye una alternativa viable».
De este modo, la Sociedad Balear de Laboratorio Clínico, Asociación Canaria del Laboratorio Clínico (ACLAC), Asociación de Analistas Clínicos de Extremadura (ANCLEX), Asociación Valenciana de Especialistas en Laboratorio Clínico (AVELAC), Sociedad Castellanomanchega de Medicina de Laboratorio (LABCAM), Sociedad Asturiana de Bioquímica y Análisis Clínicos – Medicina de Laboratorio (SAByAC ML), Sociedad Andaluza de Análisis Clínicos, Medicina del Laboratorio (SANAC) y Sociedad Castellanoleonesa de Análisis Clínicos (SCLAC) y la Sociedad de Medicina de Laboratorio de la región de Murcia (SOMELMUR), apuestan por la creación de un Diploma de Acreditación Avanzada para esta área funcional que consideran «constituye una alternativa viable».
La genética de laboratorio forma parte del núcleo competencial del Laboratorio Clínico y ha sido desarrollada durante décadas con altos estándares de calidad, responsabilidad y evidencia científica. «No se trata de una disciplina externa ni añadida, sino de una competencia estructural inherente que los especialistas ejercen con solvencia y rigor, respaldados por su trayectoria y resultados clínicos contrastados», afirman.
Las entidades firmantes consideran que cualquier revisión del mapa de especialidades sanitarias debe fundamentarse en un análisis «exhaustivo y riguroso» que evalúe sus repercusiones sobre el Laboratorio Clínico, garantizando así la preservación de la excelencia asistencial. En este sentido, apelan a la «serenidad, la cohesión y la claridad» entre los profesionales, promoviendo un diálogo constructivo sustentado en una formación sanitaria especializada en genética diagnóstica, biología molecular y medicina de precisión, pilares fundamentales para la calidad asistencial y la seguridad del paciente.
Consideran que corresponde a la Comisión Nacional de la Especialidad actualizar los programas formativos y proponer, cuando sea necesario, medidas de refuerzo utilizando las herramientas oficiales disponibles, como la ampliación del periodo formativo a 5 años o la creación de Áreas de Capacitación Específica, conforme a lo establecido en el Real Decreto 589/2022.
Las sociedades científicas consideran que la propuesta del Ministerio «no responde a carencias formativas o tecnológicas reales, sino a una problemática laboral generada por la contratación de personal no sanitario sin la formación sanitaria especializada requerida para el desempeño de funciones asistenciales». Según su experiencia, la solución no debe centrarse en la fragmentación de competencias, sino en el fortalecimiento y consolidación de las estructuras formativas actualmente vigentes.
Además, advierten que esta propuesta vulnera los principios establecidos en el Real Decreto 589/2022, normativa que dirige cualquier regulación hacia la eficiencia, la cohesión y la calidad asistencial.
«La medida no solo impide una gestión racional de los recursos públicos, sino que también fomenta la fragmentación de la formación de los profesionales y de la asistencia sanitaria, generando compartimentos estancos que comprometen la continuidad y la integración de los cuidados», afirman.
No obstante, las entidades reiteran su firme disposición a colaborar con la Administración sanitaria en el diseño de alternativas fundamentadas en la eficiencia, la evidencia científica y la coherencia estructural. Confían en que el trabajo conjunto permitirá adecuar «la oferta formativa a las necesidades reales del sistema sanitario y de la ciudadanía, evitando duplicidades y solapamientos contraproducentes».
29 septiembre 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
