
abr
21
 La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
De acuerdo con la literatura médica, es considerada poco frecuente y que aqueja a hombres y mujeres de cualquier edad, sin importar el origen étnico, e incluso puede originarse en cualquier etapa de vida del paciente desde lactantes hasta adultos.
Es una enfermedad neuromuscular hereditaria autosómica recesiva y de origen genético, causada por un gen que genera el déficit de la enzima alfa-glucosidasa ácida o maltasa ácida.
Ello permite la acumulación de una sustancia en los músculos denominada glucógeno, ocasionando pérdida de masa muscular, así como debilidad muscular en las extremidades inferiores.
Igualmente puede afectar los músculos del tronco, brazos y hombros, y la gravedad y síntomas varían en cada paciente.
Entre los malestares relacionados con esta dolencia genética están la dificultad para levantarse, al estar sentado, para caminar, subir escaleras, masticar y la debilidad muscular progresiva.
Una persona aquejada por la Enfermedad de Pompe, también puede presentar reflujo gastroesofágico, disnea, caídas frecuentes, agitación tras realizar esfuerzo físico, dolor de cabeza por las mañanas y somnolencia durante el día debido a la ortopnea.
El nombre de esta dolencia es en honor al patólogo holandés Joannes Pompe, quien presentó el primer caso en el año 1930, y cada 15 de abril es celebrado el Día Internacional de la Enfermedad de Pompe.
15 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
21
 La Secretaría de Salud (Sesal) de Honduras notificó hoy 99 casos sospechosos de gusano barrenador en humanos en lo que va del actual ejercicio.
La Secretaría de Salud (Sesal) de Honduras notificó hoy 99 casos sospechosos de gusano barrenador en humanos en lo que va del actual ejercicio.
En declaraciones a periodistas, el jefe de la Unidad de Vigilancia de la Sesal, Homer Mejía, informó que los 99 casos se encuentran en laboratorio y la mayoría de los afectados son hombres.
Esta nación centroamericana confirmó el pasado 10 de marzo el primer fallecimiento en 2026 de una persona a causa de miasis por gusano barrenador del ganado, una mujer de 78 años originaria de esta capital.
La miasis cutánea por gusano barrenador es provocada por las larvas de la mosca Cochliomyia hominivorax, que deposita sus huevos en heridas abiertas o lesiones de animales de sangre caliente, incluido los seres humanos.
Horas después de que el insecto deposita la larva, nacen los gusanos que se alimentan del tejido vivo.
Sus efectos son particularmente devastadores en la producción pecuaria, debido a la mortalidad y a un menor rendimiento en la producción de leche y carne.
Desde 1995, Honduras no registraba en humanos casos de gusano barrenador, pero en septiembre de 2024 declaró una emergencia sanitaria ante la propagación de la enfermedad parasitaria en Centroamérica y otras regiones.
Mejía exhortó a la población a cubrir cualquier herida o úlcera con gasas o vendajes para evitar que las moscas depositen sus larvas y provoquen miasis.
Recordó que el riesgo es mayor en personas de edad avanzada o con enfermedades de base, por lo que pidió extremar las medidas de prevención.
El jefe de la Unidad de Vigilancia de la Sesal indicó que la mayoría de los casos detectados se concentran en el departamento de Francisco Morazán, donde se encuentra Tegucigalpa, así como en las regiones de Olancho (oriente) y Yoro (norte).
14 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
20
 El Gobierno de Colombia trazará una hoja de ruta con el fin de consolidar estrategias y fortalecer la capacidad del país frente a emergencias y desastres durante un evento que se celebrará en mayo venidero, se conoció hoy.
El Gobierno de Colombia trazará una hoja de ruta con el fin de consolidar estrategias y fortalecer la capacidad del país frente a emergencias y desastres durante un evento que se celebrará en mayo venidero, se conoció hoy.
Según divulgó la Unidad Nacional para la Gestión del Riesgo de Desastres (Ungrd), el encuentro reunirá a más de 800 participantes entre representantes gubernamentales y territoriales, organismos de cooperación internacional, academia, sector privado y comunidades.
Comunicó la entidad que la agenda del foro, denominado Plataforma Nacional para la Gestión del Riesgo de Desastres, celebrará entre el 20 y el 22 de mayo próximo cinco plenarias, más de veinte sesiones paralelas, laboratorios de aprendizaje, entre otros.
También se prevé que los delegados analicen temas prioritarios como los sistemas de alerta temprana, el ordenamiento territorial, la infraestructura resiliente y el financiamiento del riesgo, así como el fortalecimiento de la preparación, respuesta y recuperación post desastre, con énfasis en la toma de decisiones basada en evidencia.
Adicionalmente, de acuerdo con la fuente, se abordarán enfoques relacionados con inteligencia artificial, ciencia aplicada y diálogo de saberes, junto con perspectivas diferenciales enfocadas en comunidades, género, pueblos étnicos y niñez, además del bienestar animal y el fortalecimiento de capacidades operativas ante emergencias.
La Ungrd invitó a entidades territoriales, organizaciones sociales, academia, sector privado y cooperación internacional a postular sus experiencias e iniciativas, en un esfuerzo por construir de manera colectiva las políticas públicas que orientarán la gestión del riesgo de desastres en Colombia en los próximos años.
La convocatoria del encuentro se da después de que el Instituto de Hidrología, Meteorología y Estudios Ambientales (Ideam) alertó sobre los efectos que acarrearía la llegada de El Niño al país neogranadino y pidió a las autoridades anticipar medidas de preparación y mitigación frente a posibles impactos.
El organismo advirtió que la probabilidad de que se instauren las condiciones generadas por el fenómeno climatológico se sitúa sobre el 61 % entre mayo y julio y aumenta progresivamente durante el segundo semestre hasta alcanzar índices superiores al 90 por ciento a partir de septiembre.
13 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A.| Noticia
abr
20
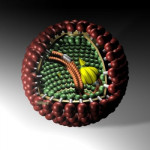 Un equipo internacional de investigadores logró descifrar la estructura del patógeno que causa esta encefalitis que casi siempre resulta fatal, por lo que el virus de Borna deja hoy de ser un verdadero misterio.
Un equipo internacional de investigadores logró descifrar la estructura del patógeno que causa esta encefalitis que casi siempre resulta fatal, por lo que el virus de Borna deja hoy de ser un verdadero misterio.
Según la revista Science Advances, los científicos hicieron varios trabajos estructurales previos sobre los complejos de nucleoproteína-ARN del virus del Ébola pudiendo profundizar en el conocimiento sobre enfermedad de Borna tipo 1 (BoDV-1).
Recuerda el artículo que los casos de la enfermedad de Borna tipo 1 (BoDV-1) son extremadamente raros en humanos, pero en quienes la desarrollan, el desenlace es grave y casi siempre resulta en encefalitis o inflamación cerebral fatal.
Este virus zoonótico pertenece al orden Mononegavirales, que incluye los virus del Ébola, el sarampión y la rabia.
El complejo nucleoproteína-ARN de estos virus protege su ARN genómico y favorece la síntesis de ARN viral, por lo que comprender la estructura de este complejo es fundamental para combatir la replicación viral.
Los investigadores completaron la caracterización estructural de varias familias de mononegavirus que infectan con mayor frecuencia a los humanos.
«Los bornavirus son menos conocidos que muchos otros virus de ARN humanos, pero representan el último caso importante sin resolver para el análisis estructural de la nucleoproteína-ARN entre los mononegavirus que infectan a los humanos», enfatizan los expertos.
El equipo se auxilió de la microscopía crioelectrónica obteniendo imágenes de alta resolución de los complejos de nucleoproteína-ARN del virus BoDV-1 y realizaron una clasificación computacional para separar y reconstruir los distintos estados de ensamblaje de cada complejo en la muestra.
Luego utilizaron ensayos mutacionales y funcionales para analizar los residuos de nucleoproteína-ARN y evaluar su función en la síntesis y el ensamblaje del ARN viral.
El estudio reveló la estructura tridimensional de este complejo, mostrando ensamblajes en forma de anillo y la unión del ARN viral en el surco interno.
También descubrieron que cada subunidad de nucleoproteína aloja ocho nucleótidos de ARN, lo que sugiere un modo de unión distinto al descrito para otros virus relacionados.
En conjunto, estos hallazgos sugieren un modelo incremental en el que el ensamblaje de nucleoproteínas y la interacción con el ARN son procesos separados pero coordinados.
13 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
17
 Una tendencia creciente de complicaciones graves en casos de varicela en adultos genera hoy preocupación en la comunidad, advirtió aquí el diario Hanoi Moi.
Una tendencia creciente de complicaciones graves en casos de varicela en adultos genera hoy preocupación en la comunidad, advirtió aquí el diario Hanoi Moi.
Cabe destacar que la mayoría de los reportes de gravedad se dan en personas no vacunadas, lo cual demuestra que la protección que brindan las vacunas aún se subestima, a pesar de ser la medida más eficaz, subrayó la publicación.
De acuerdo con la fuente, lo que antes se consideraba una enfermedad benigna en niños, en adultos puede volverse crítica rápidamente, con complicaciones como sepsis o insuficiencia multiorgánica.
El periódico señala que en la Unidad de Cuidados Intensivos del Hospital Nacional de Enfermedades Tropicales “los médicos han estado trabajando a contrarreloj para salvar la vida de muchos pacientes con complicaciones graves derivadas de la varicela”.
Según los galenos, el denominador común en la mayoría de estos casos es que los pacientes nunca han sido vacunados contra la varicela, lo cual hace que el virus sea más propenso a ataques agresivos, especialmente en personas con enfermedades preexistentes o sistemas inmunitarios debilitados.
Al respecto, la directora médica del Sistema de Vacunación VNVC, Bach Thi Chinh, citó estudios en los cuales se demuestra que la tasa de hospitalización en adultos con varicela es 13 veces mayor y la tasa de mortalidad aproximadamente 25 veces más alta que en niños.
La neumonía es la complicación más común (entre un cinco y un 15 %), con una tasa de mortalidad que puede alcanzar un 30 %.
En mujeres embarazadas el peligro es aún mayor. Aproximadamente un 20 % de los casos de varicela pueden derivar en neumonía, con una tasa de mortalidad de hasta un 40 %, y si la infección se produce entre las semanas 13 y 20 de gestación, el riesgo de defectos congénitos y muerte fetal aumenta significativamente.
Si la infección se produce en los últimos tres meses, los recién nacidos tienen un 30 % de riesgo de muerte, y alrededor de un 15 % puede desarrollar herpes zóster en sus primeros años de vida, alertó.
Más allá de los factores inmunológicos, los médicos creen que los errores en el cuidado y tratamiento en el hogar también contribuyen a la gravedad de la varicela, señaló el diario.
Hábitos como evitar bañarse y las corrientes de aire, aplicar remedios herbales, automedicarse sin receta, o apretar las ampollas, pueden dañar la barrera protectora de la piel, permitiendo la entrada de bacterias y provocando sepsis e insuficiencia multiorgánica.
12 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
17
 La tuberculosis (TB) sigue siendo hoy un importante problema de salud pública a nivel mundial, y la comunidad científica prosigue los estudios y ensayos para lograr más vacunas contra la enfermedad.
La tuberculosis (TB) sigue siendo hoy un importante problema de salud pública a nivel mundial, y la comunidad científica prosigue los estudios y ensayos para lograr más vacunas contra la enfermedad.
La vacuna BCG es actualmente la única autorizada contra la TB, y aunque es eficaz contra las formas graves en niños pequeños, no ofrece protección a adolescentes ni adultos.
El reto ahora de los científicos es lograr un inyectable que funcione en todas las edades, subraya un artículo publicado por la revista The British Medical Journal.
Un equipo de investigadores de la India evaluó dos nuevas vacunas contra la tuberculosis, denominadas VPM1002 e Immuvac.
Ambas –según la fuente- pueden proteger contra todas las formas de TB (pulmonar y extrapulmonar), prevenir la infección latente (inactiva) y generar una respuesta inmunitaria contra la bacteria que la provoca.
El Consejo Indio de Investigación Médica de Delhi aseguró que ellas para prevenir la TB son seguras en su uso en adultos y niños, pero no ofrecen protección contra todas las formas de la mencionada dolencia.
Los científicos explicaron que ninguno de los inyectables ofreció protección general contra la TB ni previno la infección latente por la misma.
Resaltaron que ambas demostraron la capacidad de prevenir la progresión a tuberculosis activa en aquellos que desarrollaron tuberculosis latente.
“Los investigadores descubrieron que, si bien ninguna de las dos vacunas demostró eficacia contra la tuberculosis total ni contra la tuberculosis pulmonar (TBP), una de ellas, la VPM1002, mostró eficacia de un 50,4 % contra la TBP en todos los grupos de edad, incluidos los de 36 a 60 años con un 79,5 %”, precisó el artículo.
Estos hallazgos –destacan los expertos- sugieren un beneficio potencialmente significativo para la salud pública, ya que la tuberculosis extrapulmonar, que afecta a órganos más allá de los pulmones, suele estar asociada a un mayor riesgo de mortalidad que la TBP.
Un hallazgo prometedor fue la protección observada contra la tuberculosis en niños de seis a menos 14 años, donde VPM1002 proporcionó protección contra todos los tipos de tuberculosis, tanto pulmonar como extrapulmonar.
En tanto, la Immuvac proporcionó protección solo contra la tuberculosis extrapulmonar en el grupo de edad de seis a menos de 10 años.
Aclaran los científicos, que ninguna de las vacunas protegió a los niños y adultos con bajo peso.
10 marzo 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
