
jun
20
 Las enfermedades gastrointestinales y respiratorias son las que más aquejan hoy en día a nuestro sistema, explicó la portavoz del Ministerio de Salud y Asistencia Social (Mspas) de Guatemala, Ana Luisa Olmedo.
Las enfermedades gastrointestinales y respiratorias son las que más aquejan hoy en día a nuestro sistema, explicó la portavoz del Ministerio de Salud y Asistencia Social (Mspas) de Guatemala, Ana Luisa Olmedo.
En relación con las primeras, la vocera de la cartera sanitaria detalló que atienden aquellas provocadas por el norovirus, rotavirus y adenovirus, que se caracterizan por diarrea, fiebre, dolor estomacal y deshidratación.
La mayor cantidad de infecciones respiratorias que se presentan son las agudas, especialmente el Virus Sincitial, agregó la funcionaria, citada por el diario local Prensa Libre.
Estas se asocian con la tendencia de las personas a estar en interiores y al hacinamiento que se da cuando tratan de protegerse de la lluvia, acotó.
En la actual temporada aumentan personas con gripe, influenza, bronquitis y neumonía, los cuales se caracterizan por síntomas como fiebre, malestar general, tos, dolor muscular y articular y congestión nasal, subrayó Olmedo.
En el caso de los niños, particularizó, afecta el virus sincitial respiratorio que causa fiebre alta, dolor de cuerpo, pérdida de apetito y decaimiento. Datos del Mspas dan cuenta de la existencia en la semana epidemiológica 22 de influenza A (H3N2), parainfluenza, virus sincitial respiratorio, adenovirus, coronavirus y metapneumovirus.
El médico internista Silvio Eduardo López confirmó al medio de prensa que las enfermedades más recurrentes, debido a las variaciones de clima, son las gastrointestinales.
Recomendó mantener una adecuada hidratación, evitar realizar actividades al aire libre, lavarse en forma correcta las manos, pero también las frutas y verduras que se consumen.
«Es importante evitar consumir alimentos en lugares donde no tengan medidas de higiene y mantenerse abrigado», enfatizó el galeno.
Ante el alza de las precipitaciones en el país, el Mspas declaró la víspera Alerta Naranja Institucional en la red de servicios de salud.
Tal condición puede cambiar –precisó- de conformidad con las condiciones, tanto a nivel nacional como para las regiones en donde el pronóstico de lluvias fue catalogado por encima de lo normal.
El organismo llamó a hervir el agua antes de consumirla, evitar el contacto con las estancadas, no nadar ni bañarse en ríos, charcas o estanques que puedan estar contaminados con residuos fecales o químicos.
Ante la necesidad de guardar el vital líquido, tapar los recipientes y deshacerse de los que puedan acumularlo, para impedir la proliferación de zancudos del dengue.
El Mspas instruyó a las Direcciones Departamentales de Redes Integradas de Servicios de Salud y Direcciones de Hospitales a tomar un grupo de medidas prioritarias.
La época de precipitaciones comenzó en Guatemala concretamente a inicios de este mes y las autoridades esperan registros históricos.
18 junio 2024|Fuente: Prensa Latina |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2023. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
jun
20
 La agencia meteorológica de Canadá advirtió hoy a las provincias del centro y este del país por una ola de calor extrema que ubicará los niveles de temperatura cercanos a los 40 °C.
La agencia meteorológica de Canadá advirtió hoy a las provincias del centro y este del país por una ola de calor extrema que ubicará los niveles de temperatura cercanos a los 40 °C.
La meteoróloga de CTV Your Morning, Kelsey McEwen, señaló que regiones de Ontario, Quebec, New Brunswick y el sur de Halifax registrarán temperaturas entre 29 y 34 grados y valores de humedad cercanos a 41, de martes a jueves.
Las autoridades animaron a los pobladores a mantenerse hidratados y tomar las medias pertinentes.
Por otra parte, Environment Canada dijo que en el noroeste de Ontario existe riesgo de tornado para hoy por la tarde.
Se pronostican tormentas eléctricas intensas para la tarde con ráfagas de viento de hasta 90 kilómetros por hora con grandes granizos y fuertes lluvias, según el aviso de la agencia.
18 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
jun
20
 El analizador de orina PA-100, que permite detectar rápidamente si una infección urinaria es causada por bacterias e identificar los antibióticos adecuados para tratarla fue galardonado con el Premio Longitude, trascendió hoy.
El analizador de orina PA-100, que permite detectar rápidamente si una infección urinaria es causada por bacterias e identificar los antibióticos adecuados para tratarla fue galardonado con el Premio Longitude, trascendió hoy.
Dicho reconocimiento, que otorga más de 10 millones de dólares, unas 10 veces lo que da el premio Nobel, recayó en esa inventiva desarrollada por expertos de la Universidad de Uppsala y comercializado por la empresa Sysmex Astrego.
Los especialistas explicaron que la prueba de orina premiada revela en 15 minutos si la infección es bacteriana y en 45 minutos qué antibióticos funcionan.
En declaraciones a la cadena británica BBC, uno de los creadores, Johan Elf, detalló que estaban trabajando en tecnología de medición cuando se lanzó el premio, hace una década, pero luego se dieron cuenta de que su trabajo «podría ser realmente útil en la lucha contra la resistencia a los antimicrobianos».
«El premio nos ayudará a adaptar la prueba para su uso con diferentes tipos de infecciones urinarias y antibióticos, acelerando el acceso para más pacientes», dijo uno de los fundadores de la compañía Sysmex Astrego, Mikael Olsson.
Los reconocimientos otorgados en el Premio Longitude sobre Resistencia a los Antimicrobianos fue anunciado en 2013 por el entonces primer ministro británico, David Cameron, y las solicitudes se recibieron a partir de mayo de 2014.
Este año, los galardones fueron para el «equipo que desarrolle pruebas de diagnóstico novedosas, asequibles, precisas y rápidas para frenar el desarrollo de resistencia a los antimicrobianos».
18 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
jun
19
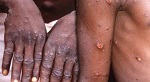 La República Democrática del Congo (RDC) registró otras 12 muertes por viruela símica (Mpox) y 427 casos sospechosos, de acuerdo con reportes de autoridades sanitarias publicados hoy por la prensa local.
La República Democrática del Congo (RDC) registró otras 12 muertes por viruela símica (Mpox) y 427 casos sospechosos, de acuerdo con reportes de autoridades sanitarias publicados hoy por la prensa local.
El jefe de operaciones del sistema de gestión de incidentes de Mpox del Programa Nacional de Lucha contra la Viruela símica y las Fiebres Hemorrágicas Virales, doctor Cris Kacita, informó que la enfermedad tiene presencia en 13 de las 26 provincias del país y Equateur continúa como la de mayor incidencia.
Durante la vigésima segunda semana epidemiológica (del 27 de mayo al 2 de junio), a la cual se refirió Kacita, se detectaron tres casos en Kinshasa, en la zona sanitaria de N’sele, dos de los cuales están hospitalizados y mantienen una buena evolución, mientras la tercera persona escapó del centro de tratamiento, según la Agencia Congoleña de Prensa.
Solo en ese periodo de tiempo Equateur registró 164 casos sospechosos y cinco muertes, por lo que siguen creciendo las nefastas estadísticas que hasta abril pasado sumaban 241 fallecidos y 3 000 casos de la enfermedad.
La RDC se encuentra actualmente entre los países más afectados por Mpox, al registrar el pasado año 14 600 casos y 654 muertes.
El país también ha disparado las alarmas de los expertos en el nivel internacional, ante la observación por primera vez de transmisiones sexuales de la enfermedad, lo que agrava la situación en materia de propagación.
La viruela símica es una enfermedad vírica rara que afecta principalmente a los primates, pero que también puede afectar a los humanos. Provoca una erupción cutánea con pústulas y llagas, así como fiebre y fatiga.
18 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
jun
19
 El acelerado aumento de casos de una rara infección bacteriana, que provoca meningitis y puede ser mortal, llevó a los funcionarios de salud de la provincia canadiense de Manitoba a emitir hoy una advertencia.
El acelerado aumento de casos de una rara infección bacteriana, que provoca meningitis y puede ser mortal, llevó a los funcionarios de salud de la provincia canadiense de Manitoba a emitir hoy una advertencia.
La región normalmente registra alrededor de seis casos al año de todo tipo de enfermedad meningocócica, pero en los últimos tiempos la cifra va por 19 personas enfermas, manifestó la doctora Carol Kurbis, directora médica de salud de Manitoba.
Agregó que otras jurisdicciones han experimentado un incremento similar, pero no están seguros de qué lo está impulsando.
«A veces vemos un aumento en este tipo de infecciones después de una pandemia o alrededor de temporadas de virus respiratorios que son altas», dijo en una entrevista en CTV Morning Live Winnipeg.
La pasada semana autoridades de salud de Toronto y Ontario notificaron también un aumento de pacientes con este tipo de afección.
La enfermedad meningocócica invasiva (EMI) es una infección bacteriana que puede causar meningitis (inflamación grave del cerebro y la médula espinal) y es potencialmente mortal.
Los síntomas de la EMI pueden variar, pero aparecen rápidamente.
Algunas personas muestran signos característicos de meningitis, con rigidez en el cuello, sarpullido violáceo y náuseas o vómitos.
Otros desarrollan síntomas de tipo sepsis, con una infección en el torrente sanguíneo.
«La gente se enferma mucho. Fiebres altas, frecuencia cardíaca rápida, dificultad para respirar y progresa bastante rápido durante el día», dijo Kurbis.
Explicó que aproximadamente una de cada diez personas porta la bacteria meningocócica en la parte posterior de la garganta y la mayoría no se enferma a causa de ella.
«Pero de vez en cuando, alguien desarrolla una enfermedad más invasiva con presentaciones como esa que pueden ser bastante graves», subrayó.
Hay dos tipos de vacunas meningocócicas. Una se dirige a la cepa B, mientras que la vacuna tetravalente se dirige a la cepa A, C, Y y W.
Se administra de forma rutinaria a los 12 meses y a los 10 años como parte del programa de inmunización escolar de la provincia.
«Seguimos las recomendaciones nacionales y hasta ahora solo estábamos vacunando a los bebés más pequeños con C, pero una vez que vimos que la cepa W estaba aumentando aquí en la provincia, cambiamos eso para asegurarnos de que cubrimos todos los infantes por cuadrivalente, también», advirtió.
17 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
jun
19
 Muchas personas con diabetes en Canadá pronto podrán administrarse insulina una vez a la semana en lugar de diariamente, anunció hoy el fabricante de medicamentos Novo Nordisk.
Muchas personas con diabetes en Canadá pronto podrán administrarse insulina una vez a la semana en lugar de diariamente, anunció hoy el fabricante de medicamentos Novo Nordisk.
La insulina icodec, que se venderá bajo la marca Awiqli, es la primera inyección de insulina basal que se administra una vez a la semana en el mundo y estará disponible en todo el país a partir del 30 de junio, dijo la compañía a The Canadian Press.
Canadá es la primera nación en recibir el producto, que fue aprobado por Health Canada en marzo para el tratamiento de adultos con diabetes tipo 1 y tipo 2.
«Creo que es algo muy importante», afirmó el doctor Harpreet Bajaj, jefe del comité directivo de directrices de práctica clínica de Diabetes Canada.
«Es enorme para reducir la carga de estas personas que necesitan inyectarse insulina», manifestó Bajaj, endocrinólogo de LMC, una clínica especializada en diabetes y endocrinología financiada con fondos públicos con ubicaciones en todo el sur de Ontario y Calgary.
Dijo que algunos de sus pacientes participaron en ensayos clínicos de Awiqli y han estado preguntando cuándo estará disponible porque han tenido que volver a las inyecciones diarias desde que terminó el estudio.
Aunque la insulina semanal cuenta con la aprobación de Health Canada para el tratamiento de la diabetes tipo 1 y tipo 2, los endocrinólogos aseguran que será más útil para los pacientes tipo 2.
Esto se debe en gran medida a que los enfermos con tipo 1 aún tendrían que aplicarse inyecciones adicionales de insulina de acción rápida a la hora de las comidas todos los días porque sus cuerpos no producen insulina por sí solos.
Las personas con diabetes tipo 2 producen insulina, pero no la suficiente o sus cuerpos no la utilizan adecuadamente.
Las inyecciones de insulina basal llevan los niveles de la hormona a la cantidad adecuada durante el ayuno, y otros medicamentos pueden controlar los «picos de azúcar que vienen con los alimentos», dijo Bajaj.
La serie de ensayos clínicos aleatorios sobre Awiqli, que incluyeron muchos países, entre ellos Canadá y Estados Unidos, se realizaron principalmente con pacientes tipo 2.
Sólo uno de los ensayos involucró a pacientes con diabetes tipo 1 y encontró un mayor riesgo de niveles bajos de azúcar en la sangre cuando tomaban la opción de insulina semanal.
La Administración de Alimentos y Medicamentos de EEUU no ha aprobado el Awiqli. Su comité asesor concluyó en mayo que se necesita más información sobre el uso de la insulina semanal en pacientes con diabetes tipo 1.
Por su parte, la Agencia de Medicamentos de Canadá, que evalúa los medicamentos y recomienda si deben calificar para reembolso bajo los planes públicos de medicamentos, estima que el costo de Awiqli será de más de 1 350 dólares al año por paciente.
La agencia recomienda en su sitio web que se financie Awiqli para el tratamiento de la diabetes tipo 2, pero con la condición de que el precio se reduzca.
17 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
