
dic
11
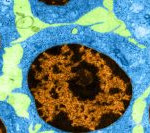 La célula leucémica corrompe la función normal de la cromatina, lo que bloquea la evolución hacia tipos celulares sanos y facilita el crecimiento tumoral, según ‘Nature Genetics’.
La célula leucémica corrompe la función normal de la cromatina, lo que bloquea la evolución hacia tipos celulares sanos y facilita el crecimiento tumoral, según ‘Nature Genetics’.
Un equipo internacional investigadores del CIMA Universidad de Navarra y de la Universidad de Cambridge ha descrito, por primera vez, los mecanismos de regulación genética que favorecen la evolución de la leucemia.
En este estudio multicéntrico, el más exhaustivo hasta el momento y que publicado en el último Nature Genetics, se han analizado las diferencias que existen en la generación de las células sanguíneas sanas frente a las células sanguíneas leucémicas.
En concreto, han caracterizado los mecanismos de regulación génica que utilizan las células para decidir cuándo y en qué medida un gen se activa o desactiva (expresión génica). Estudiar este proceso es muy importante ya que la regulación génica determina si la identidad que tomarán las células es de célula sana o de célula leucémica.
Usando tecnologías de última generación, los investigadores han desvelado que las células leucémicas corrompen mecanismos clave de regulación génica que determinan la identidad de las células sanas, lo cual bloquea su evolución hacia células sanas maduras y facilita el crecimiento del tumor.
Según el último informe de la Red Española de Registros de Cáncer (Redecan), en 2023 España contará con 6 411 nuevos casos de personas diagnosticadas de leucemia, el segundo cáncer de la sangre más prevalente. Así, este hallazgo abre la puerta al desarrollo de nuevos tratamientos para estos pacientes oncológicos.
Desregulación en el origen de la formación celular
La hematopoyesis comienza en las células madre hematopoyéticas, capaces de generar los distintos tipos de células sanguíneas (glóbulos blancos, glóbulos rojos y plaquetas). En concreto, es en la cromatina -la mezcla de ADN y proteínas que forman los cromosomas- donde se dan los procesos de regulación genética que dan lugar a la gran variedad de tipos celulares presentes en la sangre. En estos procesos intervienen dos grupos de proteínas llamados factores de la cromatina y factores de transcripción.
Los factores de transcripción marcan los genes específicos que se van a activar en cada tipo celular y los factores de la cromatina regulan la expresión de estos genes mediante cambios en la estructura bioquímica de la cromatina. De esta forma, se determina la identidad de las células sanguíneas. La desregulación de estos procesos desencadena distintos canceres sanguíneos, siendo la leucemia el segundo más frecuente.
Hasta el momento no estaba claro el papel que jugaban los factores de la cromatina en la determinación de la identidad celular. Sin embargo, este equipo de investigación ha demostrado que los factores de cromatina son un elemento crucial en la regulación de la identidad celular.
Para desmembrar esta función, han utilizado tecnologías de última generación de CRISPR y célula única.
A través del uso de tecnología que permite estudiar células individuales, hemos demostrado la complejidad de los procesos que regulan las células, revelando una gran diversidad en la función de factores de cromatina, así como otras funciones compartidas con los factores de transcripción», explican Julen Mendieta y Ainhoa Goñi, primeros autores de estudio e investigadores del Programa de Hemato-Oncología del CIMA, integrado en el Cancer Center Clínica Universidad de Navarra.
Diana ideal y específica
Estudiando los procesos que regulan la identidad celular en la leucemia, este equipo internacional ha revelado cómo las células leucémicas corrompen las funciones normales de los factores de cromatina para bloquear la evolución hacia tipos celulares sanos y facilitar el crecimiento tumoral.
En su análisis han observado que en esta alteración se formaron nuevos complejos de factores de transcripción y factores de cromatina exclusivos de las células leucémicas.
Según señala David Lara Astiaso, investigador del Departamento de Hematología de la Universidad de Cambridge y autor principal de estudio, como estos complejos son específicos de la leucemia y no son necesarios para la hematopoyesis normal, «son una diana ideal para una terapia que pueda desactivarlos sin causar ningún otro daño al paciente, a diferencia de tratamientos actuales como la quimioterapia, con altos niveles de toxicidad».
“Los fármacos epigenéticos están siendo muy útiles en ciertos linfomas y leucemias”
Una leucemia de ‘novo’ es un modelo de leucemogénesis
Nueva técnica para pronosticar la recaída en pacientes con leucemia mieloide aguda
Para el profesor Brian Huntly, director del Departamento de Hematología de la Universidad de Cambridge y codirector del estudio, identificar una nueva vía terapéutica potencial para la leucemia es especialmente importante. «Por ejemplo, en la leucemia mieloide aguda, que es la más común en adultos y muy agresiva, sólo el 15% de las personas diagnosticadas de esta enfermedad sobreviven más de cinco años».
En el desarrollo este estudio multicéntrico también ha colaborado investigadores la Universidad de Salzburgo (Austria) y de la compañía biotecnológica Relation Therapeutics (Reino Unido).
Varios de sus investigadores en España pertenecen al Centro de Investigación Biomédica en Red en Cáncer (CiberONC) y al Instituto de Investigación Sanitaria de Navarra (IdiSNA). El proyecto ha contado con la financiación de la Fundación Bancaria «la Caixa», de la Comisión Europea (Marie Skłodowska-Curie Actions) y del Cancer Research UK, Wellcome Trust, entre otras instituciones.
Ver más información: Lara-Astiaso D, Goñi-Salaverri A, Mendieta-Esteban J, Narayan N, Del Valle C, Gross T, et al. In vivo screening characterizes chromatin factor functions during normal and malignant hematopoiesis. Nat Genet [Internet].2023[citado 10 dic 2023];55, 1542:1554. https://doi.org/10.1038/s41588-023-01471-2
11 diciembre 2023 | Fuente: Diario Médico| Tomado de | Oncología
dic
8
 Un equipo internacional de científicos ha descubierto por primera vez cómo se organizan las células de un embrión para formar las extremidades humanas, lo que ayudará en el futuro a diagnosticar y a tratar las enfermedades congénitas asociadas a las mismas.
Un equipo internacional de científicos ha descubierto por primera vez cómo se organizan las células de un embrión para formar las extremidades humanas, lo que ayudará en el futuro a diagnosticar y a tratar las enfermedades congénitas asociadas a las mismas.
El hallazgo, que publica este miércoles la revista Nature, ha sido posible gracias a la aplicación de tecnologías celulares de vanguardia para crear un atlas que caracteriza el mapa celular de las extremidades humanas desde que empiezan a formarse. De este modo, los investigadores han visto cómo las extremidades se crean en una fase inicial como una especie de bolsas celulares indiferenciadas a los lados del cuerpo, sin una forma o función específicas.
Es a las ocho semanas de desarrollo cuando empiezan a estar bien diferenciadas, son anatómicamente complejas e inmediatamente reconocibles como extremidades, con dedos en las manos y en los pies. Este proceso requiere ‘una orquestación muy rápida y precisa de las células’, subraya el estudio, ya que cualquier pequeña alteración durante este proceso puede tener un efecto secundario, razón por la cual las variaciones en las extremidades se encuentran entre los síndromes más frecuentes al nacer, afectando aproximadamente a uno de cada 500 bebés en todo el mundo.
Para llegar a esta conclusión, los investigadores analizaron tejidos humanos de entre 5 y 9 semanas de desarrollo, lo que les permitió rastrear programas específicos de expresión génica, activados en determinados momentos y zonas, que dan forma a las extremidades en formación. Como parte del estudio, los investigadores demostraron que hay patrones genéticos con implicaciones en la formación de las manos y los pies, identificando ciertos genes que, cuando se alteran, se asocian a síndromes específicos de las extremidades como la braquidactilia -dedos cortos- y la polisindactilia -dedos de más o de menos-.
El equipo también pudo confirmar que muchos aspectos del desarrollo de las extremidades son comunes a humanos y ratones. En conjunto, estos hallazgos no solo proporcionan una caracterización en profundidad del desarrollo de las extremidades en humanos, sino que también aportan datos fundamentales que podrían influir en el diagnóstico y tratamiento de los síndromes congénitos de las extremidades. ‘Lo que revelamos es un proceso muy complejo y regulado con precisión. Es como ver trabajar a un escultor que va cincelando un bloque de mármol hasta revelar una obra maestra. En este caso, la naturaleza es la escultora, y el resultado es la increíble complejidad de nuestros dedos’, ha señalado uno de los autores, Hongbo Zhang, investigador de la Universidad Sun Yat-sen de Guangzhou en un comunicado.
Este estudio forma parte de la iniciativa internacional Atlas Celular Humano, cuyo objetivo es cartografiar todos los tipos celulares del cuerpo humano para transformar la comprensión de la salud y la enfermedad, y ha sido llevado a cabo por investigadores del Instituto Wellcome Sanger, el Instituto Europeo de Bioinformática (ambos en el Reino Unido), la Universidad Sun Yat-sen (China), además de colaboradores de otros centros. ‘Nuestro trabajo en el Atlas Celular Humano está profundizando nuestra comprensión de cómo se forman estructuras anatómicamente complejas, ayudándonos a descubrir los procesos genéticos y celulares que subyacen al desarrollo humano sano, con muchas implicaciones para la investigación y el tratamiento de las enfermedades’, ha señalado la autora Sarah Teichmann, doctora en el Instituto Wellcome Sanger y cofundadora del Atlas de Células Humanas.
Ver más información: Zhang B, He P, Lawrence JEG, Wang S, Tuck E, Williams BA, et al. A human embryonic limb cell atlas resolved in space and time. Nature [Internet]. 2023[citado 7 dic 2023];. https://doi.org/10.1038/s41586-023-06806-x
8 diciembre 2023|Fuente: EFE| Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2019. Agencia Informativa Latinoamericana Prensa Latina S.A.
dic
2
 Un estudio publicado en la revista ‘Nature‘ y coliderado por el Instituto de Biología Evolutiva (IBE), un centro mixto del Consejo Superior de Investigaciones Científicas (CSIC) y la Universidad Pompeu Fabra (UPF), Illumina y la Facultad de Medicina de Baylor, con la colaboración Centro Nacional de Análisis Genómico (CNAG), aporta una nueva visión sobre la información genética de los primates que podría revelar datos clave sobre las partes más desconocidas del genoma humano –el genoma no codificante–, su función en la salud y su papel en nuestra evolución.
Un estudio publicado en la revista ‘Nature‘ y coliderado por el Instituto de Biología Evolutiva (IBE), un centro mixto del Consejo Superior de Investigaciones Científicas (CSIC) y la Universidad Pompeu Fabra (UPF), Illumina y la Facultad de Medicina de Baylor, con la colaboración Centro Nacional de Análisis Genómico (CNAG), aporta una nueva visión sobre la información genética de los primates que podría revelar datos clave sobre las partes más desconocidas del genoma humano –el genoma no codificante–, su función en la salud y su papel en nuestra evolución.
Este estudio supone una continuación del número especial de ‘Science’ publicado en junio de 2023, que reunía el mayor catálogo de información genómica de primates hasta la fecha. El genoma no codificante es aquel que no contiene información sobre las proteínas del cuerpo y, a pesar de que conforma el 99 por ciento del ADN, su función se desconoce en gran medida. Gracias al ADN secuenciado en el CNAG, el estudio ha generado y comparado los genomas de 239 especies de primates y de 202 especies de mamíferos.
El análisis ha revelado que hay cientos de miles de secuencias reguladoras no codificantes, derivadas de adaptaciones evolutivas recientes, que están conservadas exclusivamente en primates y humanos. La conservación o ausencia de cambios en los elementos genómicos a lo largo de la evolución por efecto de la selección natural es un indicativo de la importancia de su función para la supervivencia de una especie o de un orden de animales como los primates, incluidos los humanos.
Una pequeña variación en su secuencia de ADN de los cientos de miles de regiones reguladoras identificadas en este estudio podría derivar en alteraciones de los rasgos biológicos humanos, incluida la salud humana. ‘La conservación en regiones del genoma humano es una de las herramientas más poderosas que tenemos para encontrar funcionalidad en el vasto genoma humano.
Entender la funcionalidad del genoma continúa siendo uno de los retos más importantes de la genética humana’, comenta Tomàs Marqués-Bonet, investigador ICREA en el IBE y catedrático de Genética del Departamento de Medicina y Ciencias de la Vida (MELIS) de la Universidad Pompeu Fabra (UPF).
UN PASO ESENCIAL PARA EL MAPEO GENÉTICO
Comprender los efectos de las variantes genéticas humanas es crucial para el diagnóstico y tratamiento precisos de las enfermedades genéticas. Sin embargo, los efectos de las variantes genéticas en el genoma no codificante siguen siendo difíciles de predecir. En cambio, con las secuencias de ADN codificantes de proteínas, una parte del genoma mucho más estudiada, se han logrado avances recientes utilizando técnicas de aprendizaje profundo o ‘Deep Learning’.
Ahora, esta tecnología podría aplicarse a las secuencias no codificantes identificadas en el estudio. ‘Mapear los elementos de secuencia conservados en el genoma no codificante constituye un paso esencial para comprender los efectos de todas las variantes en todo el genoma y vincularlos con rasgos y resultados de enfermedades específicos’, ha expresado Lukas Kuderna, primer autor del estudio, ahora investigador en Illumina y antes en la UPF.
Hasta la fecha, los estudios de genómica comparada han tenido éxito en encontrar secuencias conservadas, en especies distantes de mamíferos. Sin embargo, las adaptaciones evolutivas recientes más cercanas al origen de la especie humana han resultado mucho más difíciles de identificar.
Esto sucede porque se encuentran en el genoma no codificante que, en comparación con el ADN codificante, evoluciona mucho más rápido. Mediante la comparación de las secuencias conservadas en especies de primates y humanos, el estudio demuestra que una fracción sustancial de los elementos reguladores no codificantes del genoma humano tienen orígenes relativamente recientes. El estudio demuestra que muchos de estos elementos reguladores no codificantes, que anteriormente se pensaba que no estaban conservados y tenían un significado biológico incierto, representan en realidad adaptaciones evolutivas recientes en humanos.
‘Estos elementos reguladores del ADN, que se han conservado a lo largo de la evolución de los primates, podrían desempeñar un papel fundamental en el desarrollo de rasgos de primates y humanos, ofreciendo nuevos conocimientos sobre los fundamentos moleculares de la biología única de nuestra propia especie’, ha finalizado Marqu¨s-Bonet.
Ver más información: Kuderna LF, Ulirsch JC, Rashid S, Ameen M, Sundaram L, Hickey G, et al. Identification of constrained sequence elements across 239 primate genomes. Nature [Internet].2023.
2 diciembre 2023 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2019. Agencia Informativa Latinoamericana Prensa Latina S.A.
nov
17
 La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa que afecta a las neuronas del cerebro y la médula espinal provocando la pérdida del control muscular. Un estudio de la Universidad de Barcelona ha diseñado una potencial estrategia terapéutica para abordar esta patología que todavía no tiene tratamiento. Se trata de una trampa molecular que evita que uno de los compuestos peptídicos causantes de la ELA genética más común, el dipéptido polyGR, provoque sus efectos tóxicos en el organismo. Los resultados muestran que esta estrategia reduce la muerte de las neuronas de los pacientes y en un modelo animal (moscas del vinagre) de la enfermedad.
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa que afecta a las neuronas del cerebro y la médula espinal provocando la pérdida del control muscular. Un estudio de la Universidad de Barcelona ha diseñado una potencial estrategia terapéutica para abordar esta patología que todavía no tiene tratamiento. Se trata de una trampa molecular que evita que uno de los compuestos peptídicos causantes de la ELA genética más común, el dipéptido polyGR, provoque sus efectos tóxicos en el organismo. Los resultados muestran que esta estrategia reduce la muerte de las neuronas de los pacientes y en un modelo animal (moscas del vinagre) de la enfermedad.
Los primeros autores de esta investigación internacional publicada en la revista Science Advances son los expertos Juan Alberto Ortega Cano, de la Facultad de Medicina y Ciencias de la Salud y el Instituto de Neurociencias (UBneuro) de la UB, e Ivan Sasselli, del Centro de Física de Materiales (CSIC-UPV/EHU). También han participado investigadores de la Universidad de Zaragoza y la Northwestern University (Estados Unidos), entre otros.
Una de las causas genéticas más frecuentes de la ELA es la mutación en el gen C9orf72, ya que se encuentra en aproximadamente el 33 % de los pacientes afectados por la ELA familiar y el 5 % de los afectados por la ELA esporádica en España. En estos pacientes se generan unos dipéptidos con gran cantidad de cargas positivas que generan efectos altamente tóxicos en las neuronas motoras. En la primera parte del estudio, los investigadores combinaron técnicas computacionales y experimentales para mejorar la comprensión molecular de estos dipéptidos y cómo producen este proceso patológico.
Una unión tóxica para las neuronas
Los resultados mostraron que la toxicidad de estos compuestos se debe en parte a los que se unen al ARN ribosomal (ARNr), una molécula que participa en el proceso de traducción de la información genética y la síntesis de proteínas en la célula. «Hemos visto que estos dipéptidos, especialmente los ricos en el aminoácido arginina (poli-glicina-arginina o poly-GR), se unen a una región concreta del ARNr afectando a la biosíntesis de ribosomas (pequeñas estructuras que se encargan de sintetizar las proteínas de nuestro organismo) y la traducción de proteínas en neuronas motoras humanas, produciendo la muerte de estas», explica el profesor Juan Alberto Ortega Cano. «Además —añade el investigador— esta interacción de los poly-GR con el ARNr es mucho más fuerte que la interacción del poly-GR con otras proteínas ribosomales que se habían descrito previamente en otros estudios, y explica por qué estos dipéptidos tienen gran afinidad en unirse a los ribosomas de las células».
Ante estos resultados, los investigadores diseñaron una estrategia innovadora para engañar a los dipéptidos poly-GR y reducir su toxicidad. Crearon una trampa, una molécula que imitaba la secuencia específica del ARNr con la que se unen los poly-GR durante el proceso patológico, con el objetivo de evitar así los efectos neurotóxicos de esta unión. La aplicación de esta estrategia en neuronas derivadas de tejido de pacientes in vitro y en modelos de la enfermedad (moscas del vinagre) in vivo muestran que «reduce los defectos en la biosíntesis de ribosomas en la traducción de proteínas y la toxicidad en células que expresan poly-GR, así como la muerte en motoneuronas de pacientes de ELA con mutaciones en el gen C9orf72,», detalla el investigador.
Aunque todavía queda mucha investigación por validar y comprender completamente el funcionamiento de esta estrategia, los investigadores señalan en el artículo que estos resultados, prometedores, refuerzan la idea de que el uso de trampas de ARN es útil «no solamente para estudiar las interacciones ARN-proteína, sino también para proteger a las neuronas de los efectos perjudiciales de proteínas anómalas que se generan en otras enfermedades neurodegenerativas».
Referencia
Ortega JA, Sasselli IR, Boccitto M, Fleming AC, Forturna TR, Li Y, et al. CLIP-Seq analysis enables the design of protective ribosomal RNA bait oligonucleotides against C9ORF72 ALS/FTD poly-GR pathophysiology. Sci Adv[Internet]. 2023[citado 15 nov 2023]; 10;9(45):eadf7997. doi: 10.1126/sciadv.adf7997. Epub 2023 Nov 10.
17 noviembre 2023 | Fuente: EurekAlert| Tomado de Comunicado de Prensa
nov
7
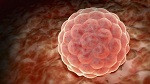 El avance, que podría ayudar en la investigación de desórdenes genéticos, pone de relieve la rapidez con la que la ciencia en este campo ha superado a la legislación.
El avance, que podría ayudar en la investigación de desórdenes genéticos, pone de relieve la rapidez con la que la ciencia en este campo ha superado a la legislación.
Un equipo de científicos ha creado embriones humanos sintéticos empleando células madre, sin la necesidad de recurrir a óvulos o esperma, un avance revolucionario que podría potencialmente ayudar en la investigación de desórdenes genéticos.
Según recoge este jueves (15.05.2023) el diario británico The Guardian, se trata de un innovador paso hacia adelante en la ciencia e investigación que plantea, sin embargo, al mismo tiempo, dilemas éticos y problemas legales.
Expertos del Reino Unido y Estados Unidos indican que estos embriones modelo, que se asemejan a los que se encuentran en las fases más tempranas del desarrollo humano, podrían proporcionar, por ejemplo, una «ventana crucial» en las causas biológicas de los abortos recurrentes.
Sin corazón latente ni cerebro
Estas estructuras no tienen un corazón latente ni el comienzo de un cerebro, aunque incluyen células que típicamente continuarían formando la placenta, el saco vitelino y el mismo embrión.
«Podemos crear modelos similares a los embriones humanos reprogramando las células», apunta la profesora Magdalena Zernicka-Goetz, del Instituto de Tecnología de California (Estados Unidos) en una intervención realizada en el marco de un congreso anual en Boston de la Sociedad Internacional para Investigación de Células Madre.
Según esto, no existe ahora la perspectiva a corto plazo de que estos embriones sintéticos vayan a ser empleados clínicamente y sería ilegal implantarlos en el útero de una paciente. Tampoco está claro todavía si estas estructuras tienen el potencial de continuar madurando más allá de las fases más tempranas de desarrollo.
La ciencia supera la legislación
De acuerdo con el medio británico, el avance pone de relieve la rapidez con la que la ciencia en este campo ha superado a la legislación, y los científicos del Reino Unido y otros países ya se están movilizando para elaborar directrices voluntarias que regulen el trabajo con embriones sintéticos.
En otra comparecencia en dicho congreso, el responsable de biología de células madre y genética del desarrollo en el Instituto Francis Crick (Londres), Robin Lovell-Badge, dijo que «la idea es que si empleamos células madre para realmente modelar el desarrollo embrionario normal humano, se puede obtener muchísima información sobre cómo comenzamos el desarrollo, lo que puede ir mal, sin tener que usar embriones en su fase temprana para la investigación».
Referencia
The Guardian. Scientists create synthetic human embryos in stem cell breakthough[Internet][citado 6 nov 2023, actualizado 2023]. Disponible en: https://www.theguardian.com/science/2023/jun/14/synthetic-human-embryos-created-in-groundbreaking-advance
7 noviembre 2023|Fuente: DW.com| Tomado de Ciencia |global
nov
3
 Una investigación puntera en terapia génica, basada en el sistema de edición genética CRISPR-Cas9, permite insertar genes con precisión, lo que ofrece esperanzas para muchos trastornos genéticos como la inmunodeficiencia combinada grave (IDCG), que afecta a los lactantes. La investigación, cuyos detalles se publican este viernes en la revista Nature Communications, ha sido realizada por científicos de la Universidad Bar-Ilan de Israel.
Una investigación puntera en terapia génica, basada en el sistema de edición genética CRISPR-Cas9, permite insertar genes con precisión, lo que ofrece esperanzas para muchos trastornos genéticos como la inmunodeficiencia combinada grave (IDCG), que afecta a los lactantes. La investigación, cuyos detalles se publican este viernes en la revista Nature Communications, ha sido realizada por científicos de la Universidad Bar-Ilan de Israel.
La IDCG es un grupo de enfermedades hereditarias del sistema inmunitario causadas por mutaciones genéticas que provocan anomalías en los linfocitos T y linfocitos B, dos tipos de glóbulos blancos necesarios para que el sistema inmune proteja al cuerpo de las infecciones. Si no se trata, la IDCG resulta mortal en el primer año de vida.
El tratamiento convencional consiste en un trasplante de sangre o de células madre hematopoyéticas (TCMH) para reconstruir el sistema sanguíneo del bebé, pero la dificultad de encontrar donantes compatibles y algunas complicaciones, como la enfermedad de injerto contra huésped (EICH), son importantes obstáculos.
Una solución innovadora ha surgido con la llegada de la edición del genoma mediante el uso de la tecnología CRISPR-Cas9, que ofrece esperanzas para muchos trastornos genéticos como la IDCG. El sistema CRISPR-Cas9 crea roturas de doble cadena específicas en el ADN, lo que permite la edición precisa de genes.
Este sistema de reparación puede alterar un gen específico o corregirlo, lo que permite atacar casi cualquier gen del genoma. Este avance abre la puerta a intervenciones terapéuticas para una amplia gama de enfermedades genómicas. El nuevo estudio presenta una prometedora técnica de edición genómica, la GE mediada por reparación homológica CRISPR-Cas9 (HDR), que ofrece la posibilidad de insertar genes con precisión. En ciertos subtipos de IDCG, una alternativa al tratamiento convencional (trasplante) puede ser la inserción genética convencional CRISPR-Cas9 mediada por HDR, pero conlleva riesgos inherentes, especialmente en los casos de IDCG causados RAG2.
RAG2 es una nucleasa que interviene en el corte del ADN durante el desarrollo de los linfocitos, y la inserción génica mediada por HDR CRISPR-Cas9 puede dar lugar a una actividad descontrolada de la nucleasa RAG2 y a variaciones estructurales perjudiciales. Para evitarlo, investigadores de la Universidad Bar-Ilan de Israel han propuesto una novedosa estrategia de sustitución, denominada ‘GE x HDR 2.0: Buscar y reemplazar’.
Este enfoque combina la edición del genoma mediada por CRISPR-Cas9 con vectores donantes de ADN adenoasociado recombinante de serotipo 6 (rAAV6) para sustituir con precisión la secuencia codificante de RAG2 y preservar los elementos reguladores. Esta estrategia puede aplicarse también a otros genes causantes de enfermedades. Para Ayal Hendel, de la Facultad de Ciencias de la Vida Goodman de la Universidad Bar-Ilan, esta técnica innovadora ‘aporta esperanza a los pacientes con RAG2-SCID y es prometedora para el tratamiento de otros trastornos genéticos’.
Referencia
Allen D, Knop O, Itkowitz B, Kalter N, Rosenberg M, Lancu O, et al. CRISPR-Cas9 engineering of the RAG2 locus via complete coding sequence replacement for therapeutic applications. Nat Commun[Internet]. 2023[citado 2 nov 2023]; 14(1): 6771. https://doi.org/10.1038/s41467-023-42036-5
2 noviembre 2023 |Fuente: EFE |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2019. Agencia Informativa Latinoamericana Prensa Latina S.A.
Noticias anteriores a 2010
Suscripción AL Día
