
may
19
El aprendizaje automático es una rama de la IA por la que los sistemas informáticos mejoran y aprenden continuamente en función de los datos y la experiencia, algo que los biólogos están utilizando para investigar las funciones centrales de los genes.
En este sentido, investigadores de la Universidad de California en San Diego (EEUU) que investigan las secuencias de ADN que activan los genes usaron inteligencia artificial para identificar una enigmática pieza del rompecabezas relacionada con la activación de genes. Este es un proceso fundamental involucrado en el crecimiento, el desarrollo y la enfermedad. Mediante aprendizaje automático, el profesor de la Facultad de Ciencias Biológicas James T. Kadonaga y su equipo descubrieron la región promotora del núcleo (DPR) aguas abajo , un código de activación de ADN de «puerta de enlace» que está involucrado en la operación de hasta un tercio de nuestros genes.
A partir de este descubrimiento, el prof.Kadonaga y los investigadores Long Vongoc y Torrey E. Rhyne han utilizado, recientemente, el aprendizaje automático para identificar secuencias de ADN «extremas sintéticas» con funciones diseñadas específicamente en la activación de genes, tal como publican en la revista ´Genes & Development´.
Los investigadores probaron millones de diferentes secuencias de ADN a través del aprendizaje automático (IA) comparando el elemento de activación del gen DPR en humanos versus moscas de la fruta ( Drosophila ). Al usar IA, pudieron encontrar secuencias DPR raras y personalizadas que están activas en humanos pero no en moscas de la fruta y viceversa. De manera más general, este enfoque ahora podría usarse para identificar secuencias de ADN sintético con actividades que podrían ser útiles en biotecnología y medicina.
«Hemos demostrado el uso de IA para el diseño de elementos de ADN personalizados en la activación de genes. Hay innumerables aplicaciones prácticas de este enfoque basado en IA. Las secuencias de ADN extremas sintéticas pueden ser muy raras, tal vez una en un millón; si existen, podrían encontrarse mediante el uso de IA. Este método debería tener aplicaciones prácticas en biotecnología e investigación biomédica», concluyó el prof. Kadonaga.
Mayo 19/2023 (IMMédico) – Tomado de Enfermedades Raras, Oncología Copyright 2023 Copyright: Publimas Digital.
may
16

Un equipo científico alemán ha desarrollado, por primera vez, modelos de ratones con un gen humano defectuoso responsable de una rara enfermedad genética: la hiperplasia suprarrenal congénita (HSC). El hallazgo, presentado en el XXV Congreso Europeo de Endocrinología que se está celebrando en Estambul, Turquía, puede ayudar a desarrollar nuevas terapias para personas con el tipo más común de esta patología congénita.
La HSC constituye un grupo de afecciones hereditarias que afecta aproximadamente a 1 de cada 15 000 nacimientos. En la forma más común, llamada deficiencia de 21-hidroxilasa, las mutaciones en el gen CYP21A2 hacen que las glándulas suprarrenales produzcan niveles bajos de cortisol y cantidades excesivas de hormonas andrógenas, como la testosterona. Este fenómeno conduce a una pubertad temprana, a que las niñas tengan rasgos masculinos y a varios problemas de salud, entre otros aspectos.
La terapia de reemplazo de hormonas esteroides es el tratamiento de primera línea, pero en muchas ocasiones puede originar efectos secundarios perjudiciales, lo que pone de manifiesto la necesidad de investigar más profundamente en la enfermedad y poder así establecer los tratamientos más efectivos y seguros.
En el hallazgo, los investigadores, del Hospital Universitario Carl Gustav Carus en Dresden, Alemania, reemplazaron el gen Cyp21a1 en ratones con el gen humano CYP21A2 portador de una mutación. Descubrieron que los ratones genéticamente modificados tenían, a las 20 semanas, glándulas suprarrenales agrandadas mientras expresaban el gen humano mutado. Además, y de manera similar a la enfermedad humana, la mutación provocó que tanto los ratones machos como las hembras tuvieran niveles bajos de corticosterona, la principal hormona del estrés en ratones que es equivalente al cortisol en humanos, y otros desequilibrios hormonales.
Aunque existen modelos animales de hiperplasia suprarrenal congénita, este es el primero en reproducir la condición humana en ratones y capaz de sobrevivir sin ningún tratamiento. «Nuestros modelos imitan con precisión los síntomas observados en pacientes humanos», según Shamini Ramkumar Thirumalasetty, coordinadora de la investigación, quien destaca que, por ejemplo, «los ratones hembra mutantes también tienen problemas de fertilidad, lo que creemos que probablemente se deba a los desequilibrios hormonales provocados por la hiperplasia suprarrenal congénita».
Este modelo de ratón abre una nueva vía que permitirá al equipo estudiar más detalladamente los mecanismos de la enfermedad y encontrar los tratamientos más efectivos.
La investigadora indica que aunque se están desarrollando nuevos tratamientos para minimizar los efectos secundarios de las hormonas esteroides, estos fármacos carecen de modelos ‘in vivo efectivos’ para las pruebas preclínicas. «Nuestro ratón puede servir como un excelente modelo para probar nuevos medicamentos y opciones de tratamiento para pacientes con hiperplasia suprarrenal congénita, como las terapias con células madre, y facilitará la transición de la investigación básica a la clínica».
Mayo 15/2023 (Diario Médico) – Tomado de Endocrinología – Presentado en el XXV Congreso Europeo de Endocrinología Copyright Junio 2018 Unidad Editorial Revistas, S.L.U. Todos los derechos reservados.
may
3
 Científicos del Centro Princesa Máxima de Oncología Pediátrica y del Instituto Hubrecht de los Países Bajos han revelado nuevos conocimientos sobre las características del carcinoma fibrolamelar (CLF), un tipo poco frecuente de cáncer de hígado infantil. Sus hallazgos, publicados hoy en Nature Communications, podrían ayudar a desarrollar nuevas terapias farmacológicas en el futuro. Los miniorganismos y el sistema de «tijeras moleculares» CRISPR-Cas9 permitieron a los investigadores comprender mejor la biología de los tumores y las consecuencias biológicas de los distintos cambios en el ADN. También descubrieron la probable célula de origen de uno de los tipos de tumor FLC.
Científicos del Centro Princesa Máxima de Oncología Pediátrica y del Instituto Hubrecht de los Países Bajos han revelado nuevos conocimientos sobre las características del carcinoma fibrolamelar (CLF), un tipo poco frecuente de cáncer de hígado infantil. Sus hallazgos, publicados hoy en Nature Communications, podrían ayudar a desarrollar nuevas terapias farmacológicas en el futuro. Los miniorganismos y el sistema de «tijeras moleculares» CRISPR-Cas9 permitieron a los investigadores comprender mejor la biología de los tumores y las consecuencias biológicas de los distintos cambios en el ADN. También descubrieron la probable célula de origen de uno de los tipos de tumor FLC.
El carcinoma fibrolamelar (FLC) es un tipo de cáncer de hígado que afecta sobre todo a adolescentes y adultos jóvenes. Afecta a una de cada 5 millones de personas al año, por lo que el carcinoma fibrolamelar puede calificarse ciertamente de raro. La tasa de supervivencia sigue siendo baja. Para cambiar esta situación, se necesitan nuevas formas de tratamiento.
Nuevos modelos organoides humanos
La Dra. Benedetta Artegiani, jefa del grupo de investigación del Centro Princesa Máxima de oncología pediátrica, y la dra. Delilah Hendriks, investigadora del Instituto Hubrecht, codirigieron un nuevo estudio sobre el carcinoma fibrolamelar utilizando tecnologías innovadoras. Esto permitió a los investigadores comprender mejor las distintas consecuencias biológicas de las diferentes mutaciones encontradas en el FLC y estudiar la biología de los tumores. Esta nueva información es necesaria para comprender por qué surgen los tumores e identificar posibles dianas para mejorar los tratamientos de la enfermedad. Artegiani explica: «En nuestra investigación utilizamos organoides de hígado humano sano, es decir, minihígados cultivados en el laboratorio. Desarrollamos una serie de organoides, todos ellos con diferentes cambios en el ADN, mutaciones que se habían relacionado anteriormente con la FLC. Cambiamos el fondo genético de los organoides utilizando la técnica de modificación del ADN CRISPR-Cas9, que funciona como una «tijera molecular». Debido a su rareza, no hay muchos tejidos tumorales disponibles para la investigación. Gracias a esta técnica pudimos estudiar este tipo de tumor».
Diferentes mutaciones genéticas subyacen a distintos grados de agresividad
Artegiani y Hendriks construyeron los modelos de organoides hepáticos modificando la proteína quinasa A (PKA) mediante CRISPR-Cas9. La PKA es una proteína de señalización compleja, capaz de activar o desactivar otras proteínas. Este «interruptor proteínico» se compone de diferentes unidades, cada una de ellas codificada por un gen diferente. Modificar la función de las distintas unidades mediante cambios genéticos parece ser crucial para la aparición de la FLC.
Los organoides contenían el denominado gen de fusión mutante DNAJB1-PRKACA. Este cambio en el ADN se encuentra con mucha frecuencia en los tumores FLC. Hendriks: «Al reconstruir esta mutación en los organoides, vimos que, efectivamente, es capaz de reflejar múltiples características de los tumores que vemos en pacientes con FLC. Sin embargo, esta única mutación causó un efecto más bien leve en el comportamiento celular y molecular general de las células hepáticas».
La situación cambió por completo cuando introdujeron otro conjunto de cambios en el ADN, también hallados en pacientes con FLC. Artegiani: «Este segundo fondo no sólo contiene una mutación en uno de los genes PKA, PRKAR2A, sino también en un gen adicional llamado BAP1. En este caso, los organoides presentaban características típicas de un cáncer agresivo. Esto sugiere que diferentes antecedentes genéticos de FLC conducen a diferentes grados de agresividad tumoral». Además, el gran efecto transformador causado por los cambios en el ADN de BAP1 y PRKAR2A permite a las células adaptarse a distintos entornos. Esto explica posiblemente el crecimiento descontrolado de las células durante la formación del tumor FLC.
Los investigadores concluyeron que, aunque las mutaciones en los genes PKA son cruciales, podrían no ser suficientes para el desarrollo de FLC. Hendriks: «Estos hallazgos abren la posibilidad de buscar otros factores que se den junto con las mutaciones de PKA en los tumores de FLC. Esto podría aprovecharse para posibles terapias futuras contra esta forma de cáncer infantil».
Descubrir la célula de origen del carcinoma fibrolamelar
Para poder desarrollar nuevas terapias, también es esencial comprender la biología del propio cáncer. Uno de los primeros pasos consiste en saber de qué tipo de célula procede el cáncer: la célula de origen. Comprender la importancia de fallos genéticos específicos en el inicio del CLF y la célula de origen podría ser crucial para entender cómo podría comportarse el tumor más adelante.
Sin embargo, durante el estudio, esto resultó especialmente difícil en el caso de la FLC. Artegiani: «La causa principal es que estos tumores presentan características tanto de los hepatocitos como de las células ductales, las dos células más importantes del hígado. Nuestros organoides mostraron que la cooperación de PRKAR2A y BAP1 transformaba un hepatocito originalmente sano en una célula ductal, con mayores características de célula madre cancerosa. Esta transformación de un tipo celular en otro se denomina transdiferenciación. Se trata de un fenómeno especialmente interesante que puede darse en diversos tumores y que dificulta especialmente la identificación de la célula de origen. Sin embargo, con el uso de nuestros modelos, pudimos descubrir los hepatocitos como la probable célula de origen».
Próximos pasos
En conjunto, este estudio supone un gran avance en la comprensión de la FLC y allana el camino para nuevas investigaciones sobre cómo tratar mejor este tipo de cáncer poco frecuente. El conocimiento de los defectos genéticos podría conducir a nuevas terapias para los niños con esta enfermedad. Y la comprensión de la importancia de fallos genéticos específicos en el inicio de la FLC también podría ayudar en el futuro a entender mejor la heterogeneidad del tumor y la respuesta entre pacientes.’
Este estudio ha sido financiado por la Fibrolamellar Cancer Foundation (FCF).
Mayo 3/2023 (EurekaAlerts!) – Tomado de News Releases – Peer-Reviewed Publication (PRINCESS MÁXIMA CENTER FOR PEDIATRIC ONCOLOGY) Copyright 2023 by the American Association for the Advancement of Science (AAAS)
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
20
En el mundo hay unos 700.000 niños con demencia infantil, causada por más de 70 enfermedades genéticas raras.
Sin embargo, una nueva investigación en la que participa el Centro de Medicina Molecular y Terapéutica Innovadora (CMMIT) ofrece esperanza a esas familias, ya que los científicos estudian si el «blanqueamiento» genético podría ser una terapia eficaz.
«Hay fármacos que se utilizan para reducir la acumulación de grasa en el cerebro que causa la demencia infantil», explicó el Dr. May Aung-Htut, que dirige la participación de Murdoch. «Sin embargo, aunque esto ayuda a paliar la afección, tiene importantes efectos secundarios. Creemos que los fármacos antisentido, o parche genético, que hemos desarrollado y aplicado con éxito a la Distrofia Muscular de Duchenne pueden ser diseñados para reducir esta acumulación de grasa con ningún o pocos efectos secundarios.»
Estos fármacos explotan la maquinaria celular para engañar a las células de que no hay mensaje génico, actuando como «apagón blanco» genético. Fueron desarrollados originalmente por los profesores Sue Fletcher y Steve Wilton del CMMIT y el Instituto Perron.
El Dr. Aung-Htut trabaja en la investigación con el profesor asociado Tony Cook de la Universidad de Tasmania y colegas del Instituto Menzies de Investigación Médica, la Facultad de Medicina de Tasmania y el Servicio de Salud de Tasmania.
«Muchas enfermedades genéticas que causan demencia infantil implican la acumulación de moléculas de grasa específicas en el interior de las células cerebrales, lo que hace que éstas se vuelvan disfuncionales y mueran», explica el profesor Cook.
La inhibición de la producción de estas moléculas de grasa mediante fármacos tradicionales ha demostrado ser prometedora para estas enfermedades en el laboratorio, pero estos fármacos tienen limitaciones y efectos secundarios que los hacen inadecuados como terapia».
«Trabajando en colaboración con la UTAS y la Universidad de Murdoch, desarrollaremos y realizaremos pruebas preclínicas de un nuevo tipo de fármaco que supere estas limitaciones, lo que esperamos que mejore la atención a los niños con demencia».
El Dr. Kris Elvidge, jefe de investigación de la Iniciativa contra la Demencia Infantil, afirmó que el proyecto ofrece esperanza a quienes padecen la enfermedad.
«Cada año nacen en Australia unos 100 bebés con una enfermedad genética que causa demencia infantil, y el 75% de los niños con demencia morirá antes de cumplir los 18 años», dijo el Dr. Elvidge. «Se necesitan desesperadamente tratamientos y curas eficaces».
Abril 17/2023 (MedicalXpress) – Tomado de Alzheimer’s disease & dementia Pediatrics https://medicalxpress.com/news/2023-04-genetic-whiteout-children-dementia.html Copyright Medical Xpress 2011 – 2023 powered by Science X Network.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
17
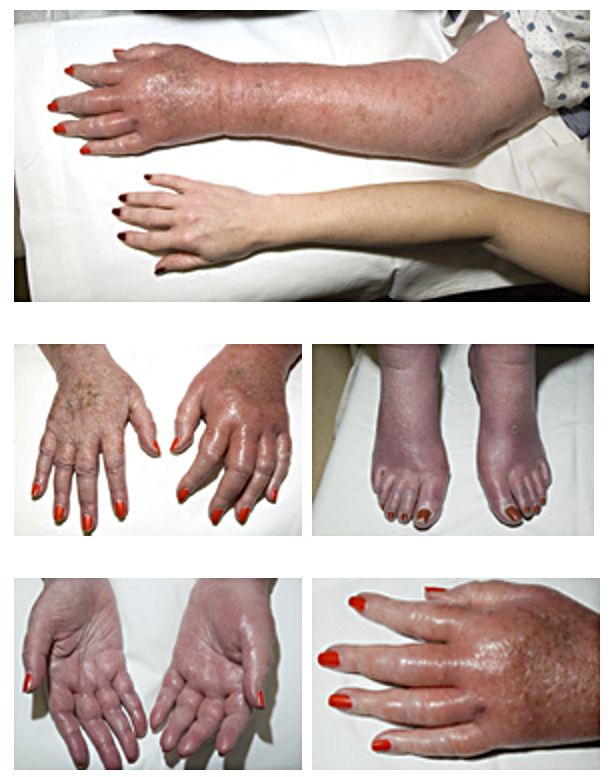 Se manifiesta como episodios de dolor ardiente asociado con pies rojos y calientes que ocasionalmente involucran las manos.
Se manifiesta como episodios de dolor ardiente asociado con pies rojos y calientes que ocasionalmente involucran las manos.
La eritromelalgia (EM) es una enfermedad debilitante rara caracterizada por episodios de dolor ardiente asociado con pies rojos y calientes y ocasionalmente las manos.
Durante los síntomas, los pies y las manos pueden estar hinchados aunque el edema no está universalmente presente o reportado.
El control endógeno de la temperatura, probablemente a través de la vasodilatación de las anastomosis arteriovenosas termorreguladoras, puede ser un factor clave en el mecanismo fisiopatológico de la eritromelalgia (EM). A menudo, se desencadenan episodios a partir de la actividad física y la exposición a temperaturas cálidas mientras que el dolor se alivia en forma característica por el enfriamiento de las zonas afectadas.
Debido a que los síntomas son intermitentes y los pies y las manos pueden parecer normales en la consulta, el diagnóstico puede ser omitido. Los pacientes suelen solicitar la atención médica de diversas especialidades, como atención primaria, dermatología, medicina vascular, neurología y medicina del dolor. Los pacientes tienen menor calidad de vida, aumento de la morbilidad y la mortalidad así como mayor riesgo de suicidio en comparación con la población general de EE. UU.
El tratamiento efectivo para el alivio de los síntomas es fundamental para evitar las complicaciones graves y mejorar los resultados. Para denominar la afección existen términos como EM primaria y EM secundaria pero su uso es variado y pueden dar lugar a confusión.
Convencionalmente, la denominación eritromelalgia secundaria se utiliza para la EM que tiene una causa subyacente identificable. El término EM primaria se utiliza para la EM sin una causa subyacente identificable (EM idiopática). También se ha utilizado el término EM primaria como sinónimo de EM heredada, para describir la EM en pacientes con antecedentes familiares de la enfermedad. Sin embargo, etiquetar la EM como primaria o secundaria no afecta mucho su manejo.
Tratamientos farmacológicos
La EM es una condición difícil de tratar. Se pueden utilizar tratamientos tópicos y sistémicos. El resultado de los tratamientos es muy variable. Muchos informes de tratamientos fueron descritos hace varios años (esta revisión bibliográfica abarca los años 1985-2021); hay pocos datos publicados que discutan la durabilidad de los tratamientos reportados.
Como tratamiento de primera línea se pueden usar medicamentos tópicos para alivio del eritema y el dolor, y quizás sea todo lo que se necesita para manejar esta condición. Para pacientes con EM secundaria por enfermedad mieloproliferativa, el tratamiento debe centrarse en abordar la causa subyacente.
La aspirina es el tratamiento sistémico de primera línea para pacientes con EM, particularmente aquellos que tienen enfermedades mieloproliferativas subyacentes. Entonces pueden considerarse otros medicamentos sistémicos.
La aplicación de un enfoque de equipo multidisciplinario es beneficioso para el cuidado de los pacientes con EM. Para pacientes que tienen EM severa, refractaria o con discapacidad, es necesaria la derivación a un centro de rehabilitación completa del dolor.
Los medicamentos tópicos pueden ser utilizados como tratamiento de primera línea para aliviar el eritema y el dolor y puede ser todo lo que se necesita para manejar esta condición. Muchos tratamientos tópicos se han utilizado para la EM, con varios niveles de eficacia. Algunos tratamientos pueden usarse para abordar principalmente el dolor asociado con la EM, mientras que otros pueden disminuir el enrojecimiento con la consiguiente disminución del dolor. Para evaluar la eficacia del medicamento tópico, debe ser utilizado durante al menos 4 semanas antes de cambiarlo por otro tratamiento.
Abril 17/2023 (IntraMed) – Tomado de Artículos. Copyright 1997-2023
abr
16
El Día Internacional del Síndrome de Wolf-Hirschhorn o también llamado 4P, se celebra el 16 de abril de cada año. Esta efeméride ha sido impulsada desde el año 2014 por asociaciones de pacientes de todo el mundo con el fin de visibilizar esta patología tan poco frecuente que afecta a un pequeño porcentaje de la población mundial y que produce trastornos genéticos significativos que inciden en su calidad de vida.
¿Qué es el Síndrome de Wolf-Hirschhorn?
Es una enfermedad genética rara causada por la pérdida de un fragmento del brazo corto del cromosoma 4 (4p), que afecta a 1 de cada 50.000 nacidos. Este trastorno se caracteriza por producir algunos cambios físicos e intelectuales en los individuos que lo padecen y que con el paso del tiempo se hacen mucho más visibles.
Las personas con este síndrome suelen manifestar síntomas de forma muy temprana. Por lo general, es más frecuente en el sexo femenino que en el masculino. Los pacientes nacen con una deformidad craneal y rasgos faciales distintivos bastante notorios. Entre las alteraciones visibles más comunes se encuentran: frente amplia con cejas demasiado arqueadas, cabeza con forma de casco y muy pequeña, orejas de tamaño reducido y formación de labio leporino. También es frecuente observar en los niños retraso en el crecimiento, problemas psicomotores, discapacidad cognitiva e intelectual, presencia de hipotonía muscular y convulsiones, entre otras.
¿Cuál es el diagnóstico, tratamiento y pronóstico de vida para estas personas?
Para dar con un diagnóstico preciso de esta enfermedad, es necesario realizar un examen físico que incluye un estudio genético molecular o citogenético. Por otro lado, el paciente será sometido a un examen electroencefalográfico, el cual arroja resultados definitivos de la presencia de esta patología hasta en un 90% de los casos.
Con respecto al tratamiento más adecuado para tratar el síndrome, por lo general, los médicos recomiendan ciertos fármacos para controlar las convulsiones, en caso de presentarse, así como terapias alimenticias y programas de terapia y rehabilitación.
El pronóstico de vida es bastante alentador, a pesar de los distintos trastornos y discapacidad que tienen estas personas. Muchos pacientes logran llegar a la edad adulta con los respectivos cuidados y atenciones que se le apliquen para mejorar su calidad de vida.
Abril 16/2023 (Asociación Española del Síndrome Wolf Hirschhorn) – Tomado de Noticias, Visibilidad. Copyright
Noticias anteriores a 2010
Suscripción AL Día
