
may
21
Conforme avanza la investigación se sabe que algunas enfermedades que siempre se han considerado raras o infrecuentes no lo son tanto. Es el caso de la amiloidosis cardiaca por transtiretina, que ya se demostró hace años que es una de las causas más frecuentes de insuficiencia cardiaca en personas mayores de 65 años.
La enfermedad se produce porque el organismo fabrica una sustancia conocida como proteína amiloide, que se deposita en forma de placas en las paredes del corazón, haciéndolas más gruesas y rígidas (por eso popularmente recibe el nombre de ‘síndrome del corazón rígido’). Esto conlleva que los pacientes sufran fatiga, retengan líquidos y tengan dificultades para hacer esfuerzos porque la insuficiencia cardiaca hace que el corazón no sea capaz de bombear la sangre que el organismo necesita.
«Este depósito de la sustancia también puede afectar al sistema eléctrico del corazón, por lo que los pacientes pueden sufrir arritmias. Si el depósito sigue progresando, el corazón va perdiendo mucha fuerza y llega un momento en el que el paciente fallece», explica Pablo García-Pavía, jefe de Cardiopatías Familiares del Hospital Universitario Puerta de Hierro e investigador del Centro Nacional de Investigaciones Cardiovasculares (CNIC) y del CIBER Cardiovascular (CIBERCV). De hecho, la supervivencia media de los pacientes sin tratamiento es de solo tres años.
La enfermedad puede tener dos orígenes: genético y asociado a la edad. El origen genético se debe a mutaciones en el gen de la transtiretina (TTR) y el amiloide puede acumularse en el corazón y en los nervios. En este caso, los pacientes tienen dificultades para caminar, moverse, etc. Por otro lado, cuando está asociado a la edad solo hay afectación del corazón, pero no hay síntomas neurológicos. Esta forma es mucho más frecuente, «abarca aproximadamente de un 90 a 95% de los casos, por cuanto las formas hereditarias abarcan el 5-10% restante.
Tratamientos solo para frenar la progresión de la enfermedad
Hasta hace pocos años no había ningún tipo de tratamiento para la amiloidosis cardiaca por transtiretina. En 2019 se aprobó en Europa un fármaco oral contra la enfermedad, el tafamidis, que es un estabilizador de la transtiretina. «Lo que hace es impedir el depósito del amiloide tanto en el corazón, como en los nervios (cuando esto sucede). Es decir, enlentece la progresión de la enfermedad», indica García-Pavía.
Según García-Pavía , aunque este fármaco frena la progresión de la enfermedad no mejora el estado de los pacientes porque no elimina el amiloide que ya hay depositado en el corazón. Es ahí donde entra este nuevo tratamiento cuyos abren una puerta a la esperanza para estos pacientes.
El estudio se centra en la capacidad del organismo para desarrollar un mecanismo natural para eliminar el amiloide ya acumulado. Este nuevo fármaco “lo que busca es acelerar ese sistema natural de retirada del amiloide», señala García-Pavía. Para ello, el especialista explica que el laboratorio suizo Neuroimmune, ha analizado “a personas muy mayores y sanas, ha buscado qué anticuerpos tenían en su sangre y ha localizado uno que tiene la capacidad de unirse al amiloide por transtiretina».
Ese anticuerpo se emplea para estimular el sistema defensivo de los pacientes y a su vez, para eliminar el amiloide pegado a las paredes del corazón. «El laboratorio suizo lo ha seleccionado y producido a gran escala. Es como utilizar la sabiduría natural del organismo de las personas mayores para fabricar un medicamento que sirva a la gente enferma».
El fármaco NI006, como se llama por ahora, se administró en el ensayo por vía intravenosa a dosis progresivamente mayores de forma mensual durante un año. «Los pacientes que recibieron más dosis aparentemente tuvieron una mayor reducción de los depósitos de amiloide en el corazón y mejoría de diversos parámetros cardiacos. El estudio abre la puerta a una nueva generación de fármacos destinados a una enfermedad que se ve que cada vez es más frecuente y con el progresivo envejecimiento de la población estará cada vez más presente», remata García-Pavía.
Para Pavía, hay cuatros factores que van a hacer que se diagnostique cada vez más esta enfermedad: «La población cada vez está más envejecida y al estar asociada a la edad, cuanto más vivamos más pacientes van a padecerla. En segundo lugar, que se conoce más, que haya más conocimiento hace que se diagnostiquen más pacientes. En tercer lugar, han mejorados las técnicas diagnósticas: antes esta enfermedad solo podía diagnosticarse mediante una biopsia de corazón, mientras que ahora existen técnicas de imagen que permiten detectar el depósito de amiloides sin biopsia. En cuarto lugar, la disponibilidad de tratamientos, que hacen que aprendamos más de la enfermedad y busquemos más los pacientes porque les podemos ofrecer algo».
40 pacientes de seis países
Este primer ensayo se ha hecho en 40 pacientes de seis hospitales europeos (tres de Francia, uno de Alemania, uno de Holanda y el Puerta de Hierro de España). Como todo estudio fase I, se ha analizado el perfil de seguridad del fármaco y a qué dosis puede ser efectivo. A pesar de los buenos resultados, García-Pavía es prudente y recalca que se necesitan más estudios con mayor número de pacientes. «Constituye un avance importante ya que no había ningún fármaco para esta enfermedad que mejorase al paciente y le quitara el amiloide del corazón, pero es necesario seguir haciendo pruebas.”
El especialista incide en esa cautela: «Hemos visto que la medicación es segura y que las dosis de más de 10 miligramos por kilo de peso del medicamente parecen efectivas para quitar el amiloide, pero tendremos que hacer un estudio con muchos más pacientes, entre 600 y 1.000, y una dosis que se considere efectiva, que será más de esos 10 miligramos por kilo de peso, para ver si se confirma esto que hemos visto en el ensayo fase I. Si se demuestra, ya se solicitaría la aprobación para que acabe llegando a los hospitales. Pero en este momento no creo que la gente tenga que ver esto aún como la cura de la enfermedad».
Ese estudio fase III en el que se probará la medicación frente a placebo en un número importante de personas comenzará después del verano. Ahí ya participarán países de todo el mundo, incluyendo EEUU. De hecho, la farmacéutica estadounidense Alexion, la división de enfermedades raras de AstraZeneca, ha comprado la molécula para realizar esta fase III y comercializarla en caso de que se demuestre su efectividad y se apruebe.
Mayo 21/2023 (Diario Médico) – Tomado de Cardiología – Participan seis hospitales europeos liderados por el Puerta de Hierro Copyright Junio 2018 Unidad Editorial Revistas, S.L.U.
may
10
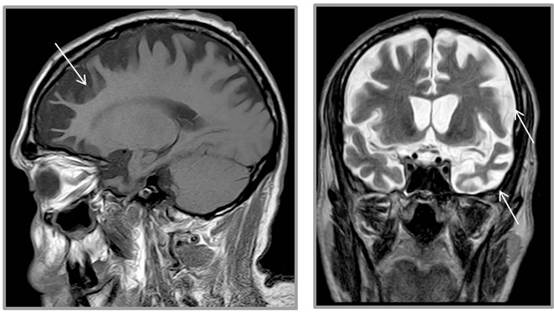
Una nueva investigación ha descubierto que algunos pacientes con enfermedad de la motoneurona (EMN) y demencia frontotemporal (DFT) son portadores de los mismos defectos genéticos raros que causan otras enfermedades neurodegenerativas.
Investigadores del Centro de Investigación de la MND de la Universidad Macquarie y del Instituto de Investigación Médica Walter y Eliza Hall han identificado los defectos en los genomas de algunas personas con MND y FTD no hereditarias o esporádicas.
La EMN provoca la muerte de las neuronas, o nervios motores, que conectan el cerebro y la médula espinal con los músculos. Son las células que controlan nuestra capacidad para movernos, respirar y tragar. La enfermedad es progresiva y finalmente mortal.
La FTD también causa la muerte de neuronas en parte del cerebro, lo que provoca una serie de síntomas progresivos como pérdida de memoria, comportamiento inusual, cambios de personalidad y problemas de comunicación. Es la misma forma de demencia que se le diagnosticó recientemente al actor Bruce Willis y, a diferencia de la demencia de inicio más antiguo, suele afectar a personas menores de 65 años.
La mayoría de los casos de ambas enfermedades –alrededor del 90% en el caso de la MND y del 60%-70% en la FTD– son esporádicos, y el resto se dan en familias.
Estos defectos genéticos, conocidos como expansiones cortas de repeticiones en tándem, son la causa de más de 20 enfermedades neurodegenerativas, entre ellas las ataxias espinocerebelosas y la distrofia miotónica. Este estudio australiano ha sido la evaluación más completa de estos defectos genéticos en pacientes con EMN y FTD de todo el mundo.
La Dra. Lyndal Henden, investigadora postdoctoral de la Universidad Macquarie, afirma que los resultados fueron una sorpresa.
«Descubrimos que casi el 18% de los pacientes esporádicos de EMN y FTD eran portadores de una expansión repetida del ADN que se cree que está implicada en otras enfermedades degenerativas», afirma.
«Descubrir esta conexión genética entre la EMN y la FTD ofrece una nueva oportunidad de descubrir factores de riesgo comunes para la muerte neuronal, y tendrá implicaciones para comprender ambas enfermedades».
La profesora asociada de la Universidad Macquarie Kelly Williams dirigió el estudio y afirma que el equipo sospechaba que podría haber cierto solapamiento con otras enfermedades, pero no hasta tal punto.
«Esto sugiere factores de riesgo compartidos entre estas enfermedades, mecanismos compartidos que provocan la muerte de los nervios, y quizá estrategias terapéuticas compartidas en el futuro», afirma.
«Aunque las causas de la EMN esporádica y la FTD siguen siendo desconocidas, éste es un paso importante en un esfuerzo a largo plazo para identificar los factores de riesgo de desarrollar una de estas enfermedades».
Ahora se puede empezar a trabajar para entender cómo estas expansiones repetidas compartidas contribuyen a la muerte neuronal.
El estudio, publicado en el último número de la revista Science Advances, es la culminación de 10 años de investigación que no habrían sido posibles sin la colaboración de pacientes con EMN y FTD, que han donado muestras biológicas para ADN tanto en la Universidad Macquarie como en la Universidad de Sídney.
Mayo 10/2023 (MedicalXpress) – Tomado de Genetics – Neuroscience Copyright Medical Xpress 2011 – 2023 powered by Science X Network.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
may
3

Un equipo de 87 investigadores de varias instituciones dirigido por la Facultad de Salud Pública de la Universidad de Michigan ha publicado un artículo en Science Advances titulado «The genetic determinants of recurrent somatic mutations in 43,693 blood genomes» (Los determinantes genéticos de las mutaciones somáticas recurrentes en 43.693 genomas sanguíneos), que cuestiona algunos supuestos largamente sostenidos sobre las mutaciones somáticas no oncogénicas.
El grupo analizó 43.693 genomas completos de muestras de sangre de 37 cohortes e identificó 7.131 mutaciones somáticas sin sentido recurrentes en al menos 50 individuos. Según los investigadores, estas mutaciones somáticas recurrentes sin sentido (RNMSM) no se explican claramente por otros fenómenos clonales, como la hematopoyesis clonal.
Las mutaciones somáticas son ligeras variantes en los genes que se adquieren en vida después del nacimiento. Son comunes en todos los animales y suelen suponer un riesgo patológico bajo a menos que se produzcan en una proteína funcional. Las mutaciones somáticas son, por definición, mutaciones que no afectan a las células sexuales, por lo que no son heredables.
Se observó que la prevalencia de las RNMSM aumenta con la edad, como cabría esperar, ya que una persona de 50 años de media tiene 27 RNMSM. Lo que puede no esperarse es una variación hereditaria de la línea germinal asociada a la adquisición de RNMSM. Estas variantes se encontraron en genes implicados en la función inmunitaria adaptativa, la producción de citocinas proinflamatorias y el compromiso de linaje linfoide. Los investigadores también hallaron ocho RNMSM asociadas a rasgos de las células sanguíneas con tamaños de efecto comparables a los de las mutaciones genéticas mendelianas.
Los autores afirman que se cree que las mutaciones somáticas no oncogénicas son infrecuentes e intrascendentes, pues tienen poco que ver con la función celular, ya que no alteran las proteínas ni causan patologías. Sin embargo, los investigadores descubrieron que las mutaciones somáticas en la sangre son un fenómeno inesperadamente común, con determinantes específicos según la ascendencia y consecuencias para la salud humana.
Los sitios de variantes no codificantes asociados positivamente a los rasgos de las células sanguíneas son especialmente interesantes. Dado que no se cree que contribuyan a los rasgos o la patología, estos cambios han pasado desapercibidos y, por tanto, no se ha estudiado su posible relación con la manifestación de enfermedades.
Los hallazgos de los autores sobre una clase de variación genética, ampliamente presente en todo el genoma humano, con determinantes genéticos de la línea germinal y consecuencias fenotípicas refutan directamente lo que antes se suponía pero nunca se había estudiado.
Mayo 3/2023 (MedicalXpress) – Tomado de Genetics Copyright Medical Xpress 2011 – 2023 powered by Science X Network
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
23
Alrededor de 170 millones de personas en todo el mundo padecen degeneración macular asociada a la edad (DMAE), una enfermedad ocular progresiva que afecta a la visión de las personas.
La DMAE avanzada se considera la principal causa de discapacidad visual y ceguera irreversible en todo el mundo.
De los dos tipos de DMAE -húmeda y seca-, alrededor del 15% de todos los casos de DMAE son de DMAE húmeda, que es la forma más grave de la enfermedad.
El estudio se ha publicado recientemente en The Journal of Clinical Investigation.
¿Qué es la DMAE húmeda?
La DMAE se produce cuando la mácula del ojo -que forma parte de la retina que controla la visión en línea recta- se daña. El daño puede deberse al envejecimiento o a factores genéticos o ambientales.
La mayoría de las personas con DMAE padecen DMAE seca, en la que se forman depósitos en la retina denominados drusas, que afectan a la visión.
La DMAE húmeda se produce cuando empiezan a crecer vasos sanguíneos anormales en la parte posterior del ojo. Estos vasos pueden filtrar lípidos, dañando la mácula y la retina.
Los síntomas de la DMAE húmeda son:
- visión borrosa
- visión central reducida
- líneas rectas que aparecen onduladas o dobladas
- zonas oscuras o ciegas en la visión
- necesidad de luz más intensa para leer
Los factores de riesgo para desarrollar DMAE incluyen
- antecedentes familiares
- edad: las personas de 55 años o más tienen un riesgo mayor
- tabaquismo
- obesidad
- hipertensión
- mala alimentación
En la actualidad no existen terapias que puedan revertir ninguno de los dos tipos de DMAE. Sin embargo, los médicos pueden ayudar a ralentizar la progresión de la enfermedad.
Comparación de los tratamientos de la DMAE húmeda
Según el Dr. Akrit Sodhi, profesor asociado de oftalmología del Wilmer Eye Institute y autor principal de este estudio retrospectivo, los investigadores decidieron centrarse en el aflibercept y el bevacizumab, ya que son los dos fármacos más utilizados para el tratamiento de las personas con DMAE húmeda.
«La decisión de elegir uno sobre otro sigue siendo un tema de debate», dijo Sodhi a Medical News Today. «Queríamos saber si la elección de uno de estos dos fármacos influía en el éxito del destete de los pacientes con DMAE húmeda de la terapia anti-VEGFFFuente de confianza».
Para este estudio, Sodhi y su equipo examinaron los datos de 106 personas con DMAE húmeda tratadas en el Instituto Oftalmológico Wilmer. Cada persona recibió una inyección mensual durante los tres primeros meses de tratamiento con aflibercept o bevacizumab.
En cada visita de seguimiento, si la DMAE húmeda de una persona se consideraba inactiva, el tiempo entre visitas se prolongaba dos semanas. Si ese intervalo podía aumentarse con seguridad a 12 semanas, se interrumpía el tratamiento y la persona era controlada por médicos sin tratamiento.
Las personas que mantuvieron una visión estable y no necesitaron una inyección ocular durante al menos 30 semanas se consideraron «destetadas» del tratamiento.
Tras el análisis, el equipo de investigación informó de que el 50% de las personas tratadas con aflibercept se consideraron retiradas del tratamiento tras un periodo de un año, en comparación con el 27% de las personas tratadas con bevacizumab.
En el caso de las personas que seguían necesitando inyecciones, los intervalos entre ellas eran un 44% más largos en las tratadas con aflibercept que en las tratadas con bevacizumab.
«Estudios anteriores han sugerido que el aflibercept puede tener algunas ventajas sobre el bevacizumab para el tratamiento de la DMAE húmeda», dijo Sodhi. «En comparación con el bevacizumab, el aflibercept dura más -lo que permite intervalos más largos entre tratamientos- y parece ser más eficaz para reducir el líquido cuando se utiliza el mismo intervalo de tratamiento.»
«Sin embargo, estas diferencias son modestas y aún se debate si justifican el coste entre 10 y 20 veces superior de aflibercept en comparación con bevacizumab», añadió.
Próximos pasos de la investigación
Medical News Today también habló con el Dr. Thomas Hanscom, oftalmólogo y especialista en retina del Providence Saint John’s Health Center de California, que no participó en esta investigación.
Dijo que está totalmente de acuerdo con el punto subyacente de la investigación, que es que aflibercept es un fármaco superior en comparación con bevacizumab.
«Se trata de una batalla antigua, porque tanto el bevacizumab como el aflibercept llevan 10 años en el mercado», afirma Hanscom. «El siguiente paso es realmente una mayor recopilación de datos sobre la próxima generación. Hay un nuevo fármaco llamado Vabysmo, que es FaricimabTrusted Source fabricado por Genentech, que ya ha demostrado tener una duración de efecto superior al aflibercept.»
Sin embargo, expresó su preocupación por algunas cuestiones estructurales del estudio y por el término «destete».
«Destete suena bien – eso es lo que todo el mundo quiere, que la gente deje de recibir estas inyecciones», explicó Hanscom. «Si nos fijamos en las demás publicaciones importantes sobre esta enfermedad, nadie utiliza nunca la palabra destete».
«En un párrafo (los autores del estudio) definen el destete como no necesitar tratamiento durante 30 semanas; eso no es realmente destete», continuó. «Eso es decir que me voy a inyectar cada 30 semanas, que es mejor que cada dos meses. Así que la definición del término destete es vaga».
«Hay una minoría -el 10 por ciento o el 20 por ciento- que al cabo de los años, sobre todo cuando el paciente llega a una edad muy avanzada, normalmente más de 90 años, algunas de esas personas parecen necesitar muchas menos inyecciones», añadió Hanscom. «Pero decir que el 43 por ciento de las personas en aflibercept puede ser destetado está muy fuera de la literatura normal».
Abril 21/2023 (MedicalNewsToday) – Tomado de Health News https://www.medicalnewstoday.com/articles/one-of-two-widely-used-macular-degeneration-drugs-outperforming-other-at-weaning-patients-off-treatment-at-one-year Copyright 2023 Healthline Media UK Ltd, Brighton, UK.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
23
En los últimos años se han producido avances notables en la capacidad para analizar e interpretar el genoma humano. No es de extrañar por lo tanto, que la realización de pruebas genéticas se haya estandarizado como estrategia diagnóstica para diferentes tipos de enfermedades.
Una consecuencia de conocer cada vez mejor el genoma humano es que puede ocurrir que se solicite una prueba genética con un objetivo clínico y se obtengan resultados con posible impacto fuera de ese objetivo. Por ejemplo, puede suceder que durante una evaluación diagnóstica se identifique en el paciente la presencia de una variante genética de riesgo para una enfermedad cardiovascular, para la que el paciente no muestra ningún síntoma. Este tipo de resultados secundarios plantea un nuevo escenario para los profesionales sanitarios, en el que deben evaluar las variantes genéticas identificadas y su posible papel en el contexto clínico de sus pacientes.
Con el objetivo de guiar a los profesionales clínicos en cómo incorporar los resultados secundarios relacionados con enfermedades cardiovasculares hereditarias en la evaluación y cuidado de sus pacientes, la Asociación Americana del Corazón ha publicado un artículo con recomendaciones y pautas para interpretar las variantes relacionadas con este tipo de enfermedades. El trabajo se ha publicado en Circulation: Genomic and Precision Medicine.
Resultados secundarios en enfermedades cardiacas hereditarias
El artículo está enfocado específicamente en la identificación de variantes genéticas en genes relacionados con enfermedades cardiacas hereditarias como canalopatías, cardiomiopatías, enfermedad torácica aórtica, dislipidemias y enfermedad cardiaca congénita.
En el artículo, los investigadores incluyen una lista de los genes relacionados con enfermedades cardiovasculares considerados por el Colegio Americano de Genética Médica como genes accionables (genes cuya alteración puede tener un impacto importante importante para la salud y para los que existen estrategias terapéuticas para prevenir o reducir su efecto dañino.). La identificación de variantes secundarias en estos genes puede tener relevancia clínica para los pacientes por lo que habría que considerar su evaluación.
“La lista de variantes incidentales relacionadas con la enfermedad cardiovascular continúa evolucionando”, ha destacado Landstrom, profesor de Pediatría y Biología Celular en la Universidad Duke y presidente del comité que ha redactado el informe. “Este documento proporciona una base de cuidado que podría ayudar a las personas con una variante genética relacionada con enfermedad cardiovascular y a sus profesionales de la salud a dar el siguiente paso en la determinación del riesgo individual y familiar que la variante puede o no tener”.
Puntos clave para interpretar las variantes incidentales
El informe, primero en ofrecer recomendaciones específicas sobre las variantes genéticas secundarias en el ámbito de las enfermedades cardiovasculares hereditarias, destaca los siguientes puntos en la interpretación de variantes:
Únicamente deberían comunicarse variantes incidentales asociadas con enfermedades cardiovasculares (de riesgo de patogenicidad o de bajo riesgo de patogenicidad) al paciente que ha aceptado conocer estos resultados antes de que la prueba genética se haya realizado.
La interpretación inicial de una variante como asociada a enfermedades cardiovasculares no siempre es precisa y puede cambiar a lo largo del tiempo.
Debería establecerse una pauta de trabajo para la interpretación de variantes incidentales en genes relacionados con enfermedades cardiovasculares que incluya: una evaluación exhaustiva específica de la enfermedad en cuestión para el paciente y una reevaluación de la asociación con la variante o el gen con la enfermedad cardiovascular en cuestión para llegar a la determinación de un riesgo de la variante a estar asociada al desarrollo de la enfermedad cardiovascular.
Esta pauta de trabajo de evaluación del riesgo de variantes determina el manejo clínico apropiado y el seguimiento del paciente y su familia.
La pauta de trabajo para la interpretación de variantes incidentales en genes relacionados con enfermedades cardiovasculares debería realizarse en un centro especializado en enfermedades cardiovasculares a través de una aproximación basada en equipos multidisciplinares.
En todo este proceso, los autores destacan la importancia de trabajar en equipos multidisciplinares que puedan “optimizar la evaluación del paciente y proporcionar apoyo continuo tanto para el paciente como para el seguimiento genético” en un entorno que evoluciona de forma tan rápida como la genética cardiovascular.
Fuente: What to do when a genetic test result signals possible heart risk. https://www.heart.org/en/news/2023/03/27/what-to-do-when-a-genetic-test-result-signals-possible-heart-risk
Abril 23/2023 (Genotipia) – Tomado de Genética Médica News https://genotipia.com/genetica_medica_news/prueba-genetica-enfermedad-cardiaca/ Copyright 2023 Genotipia.
abr
20
En el mundo hay unos 700.000 niños con demencia infantil, causada por más de 70 enfermedades genéticas raras.
Sin embargo, una nueva investigación en la que participa el Centro de Medicina Molecular y Terapéutica Innovadora (CMMIT) ofrece esperanza a esas familias, ya que los científicos estudian si el «blanqueamiento» genético podría ser una terapia eficaz.
«Hay fármacos que se utilizan para reducir la acumulación de grasa en el cerebro que causa la demencia infantil», explicó el Dr. May Aung-Htut, que dirige la participación de Murdoch. «Sin embargo, aunque esto ayuda a paliar la afección, tiene importantes efectos secundarios. Creemos que los fármacos antisentido, o parche genético, que hemos desarrollado y aplicado con éxito a la Distrofia Muscular de Duchenne pueden ser diseñados para reducir esta acumulación de grasa con ningún o pocos efectos secundarios.»
Estos fármacos explotan la maquinaria celular para engañar a las células de que no hay mensaje génico, actuando como «apagón blanco» genético. Fueron desarrollados originalmente por los profesores Sue Fletcher y Steve Wilton del CMMIT y el Instituto Perron.
El Dr. Aung-Htut trabaja en la investigación con el profesor asociado Tony Cook de la Universidad de Tasmania y colegas del Instituto Menzies de Investigación Médica, la Facultad de Medicina de Tasmania y el Servicio de Salud de Tasmania.
«Muchas enfermedades genéticas que causan demencia infantil implican la acumulación de moléculas de grasa específicas en el interior de las células cerebrales, lo que hace que éstas se vuelvan disfuncionales y mueran», explica el profesor Cook.
La inhibición de la producción de estas moléculas de grasa mediante fármacos tradicionales ha demostrado ser prometedora para estas enfermedades en el laboratorio, pero estos fármacos tienen limitaciones y efectos secundarios que los hacen inadecuados como terapia».
«Trabajando en colaboración con la UTAS y la Universidad de Murdoch, desarrollaremos y realizaremos pruebas preclínicas de un nuevo tipo de fármaco que supere estas limitaciones, lo que esperamos que mejore la atención a los niños con demencia».
El Dr. Kris Elvidge, jefe de investigación de la Iniciativa contra la Demencia Infantil, afirmó que el proyecto ofrece esperanza a quienes padecen la enfermedad.
«Cada año nacen en Australia unos 100 bebés con una enfermedad genética que causa demencia infantil, y el 75% de los niños con demencia morirá antes de cumplir los 18 años», dijo el Dr. Elvidge. «Se necesitan desesperadamente tratamientos y curas eficaces».
Abril 17/2023 (MedicalXpress) – Tomado de Alzheimer’s disease & dementia Pediatrics https://medicalxpress.com/news/2023-04-genetic-whiteout-children-dementia.html Copyright Medical Xpress 2011 – 2023 powered by Science X Network.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
Noticias anteriores a 2010
Suscripción AL Día
