
may
16

Hace cuatro años un grupo de investigadores encontró a una mujer que podría tener la llave contra la demencia más común en el mundo, el alzhéimer. Aquella huella genética se suma ahora a otra que han hallado en un hombre de 67 años y que servirá como nueva pista con la enfermedad que afecta a más de 38 millones de personas en todo el mundo.
En esta ocasión, la pieza del puzle que porta el hombre está en el gen RELN, H3447R (denominado Reelin-Colbos por el estudio de investigación de biomarcadores de Colombia-Boston), que codifica la proteína de señalización reelina, clave en las principales alteraciones cognitivas y bioquímicas de las demencias relacionadas con la proteína tau. En el caso de la mujer, el misterio se está en una mutación rara en el gen APOE3, uno de los responsables del desarrollo de la neurodegeneración, que actúa como freno frente a la aparición de los síntomas asociados con la enfermedad de Alzheimer. «Hemos caracterizado el segundo caso del mundo en el que hemos comprobado la resiliencia frente la enfermedad de Alzheimer autosómica dominante (ADAD)», explican los autores en Nature.
El equipo de investigación que ha dado con el varón y la nueva mutación es el mismo que encontró a la mujer. El grupo multidisciplinar de la Facultad de Medicina de la Universidad de Harvard, Centro Médico Universitario de Hamburgo-Eppendorf y el Grupo de Neurociencias de la Facultad de Medicina de la Universidad de Antioquia en Medellín (Colombia) llevan décadas resolviendo el rompecabezas de las mutaciones genéticas que causan Alzheimer entre los habitantes emparentados en una región rural de Antioquía y que provocan la aparición de síntomas a edades muy tempranas.
Para poner en contexto ambos casos, Joseph F. Arboleda-Velasquez, uno de los autores y profesor asociado Facultad de Medicina de Harvard, explica a El Mundo que «si en la mujer protegida la mutación estaba en el gen APOE, ahora en el varón la mutación se encuentra en el gen Reelin». Y continúa aportando detalles de las diferencias en cada caso. «En ella, la mutación de APOE se denominó Christchurch, las pruebas mostraban un cerebro que tenía muchas placas de amiloide, pero poca acumulación de tau, otra proteína que también resulta importante para en el alzhéimer. En el hombre se ha observado, además, una elevada presencia de ambas proteínas, tanto amiloide como tau, lo cual indica que es posible estar protegido incluso cuando la patología clínica de alzhéimer es alta».
La ADAD es una forma hereditaria rara de la demencia y que está más comúnmente causada por mutaciones específicas en el gen PSEN1-E280A que codifica la proteína transmembrana presenilina 1. «Se caracteriza por la aparición temprana de deterioro cognitivo, como déficits de memoria, a una edad temprana, típicamente a los 40-50 años de edad», explican los investigadores.
En la publicación se pone en contexto la situación del hombre frente a la de su hermana que sí que desarrolló la sintomatología clínica del alzhéimer y terminó falleciendo a los 73 años, con un cúmulo de patologías asociadas, tras iniciar la demencia a los 61 años. Ambos compartían la mutación PSEN1-E280A con la variante Reelin. Ella también presentó un retraso en la edad de inicio del deterioro cognitivo, aunque con una protección menor en comparación con su hermano y una enfermedad en etapa terminal prolongada. «La hermana tenía antecedentes de una lesión grave en la cabeza, que requirió cirugía reconstructiva, así como antecedentes de depresión e hipotiroidismo. Estos factores pueden haber tenido un impacto en su perfil clínico y deben tenerse en cuenta al evaluar su fenotipo», puntualizan en las conclusiones los autores.
Los investigadores explican que compararon al varón con la mujer, los dos con ADAD tardía. «Ambas personas mostraron una patología amiloide generalizada y considerable en el cerebro, que es un sello patológico de la enfermedad de Alzheimer». Y apuntan las diferencias encontradas: «Sin embargo, hubo una agregación limitada de tau (una proteína estabilizadora de microtúbulos en el cerebro) en la corteza entorrinal, una región del cerebro que se ve afectada característicamente en las primeras etapas clínicas de la enfermedad de Alzheimer».
Para verificar qué sucedía los autores realizaron la secuenciación genética y encontraron que el varón albergaba un tipo diferente de mutación: una nueva variante rara de RELN (H3447R denominada Reelin-COLBOS). Los investigadores detallan los complejos procesos que sirven para explicar la resistencia al desarrollo de las manifestaciones de la neurodegeneración que «esta mutación da como resultado un ligando RELN, una molécula de unión, que puede ser más eficaz para limitar la agregación de tau, pero para lo que se necesita más investigación». Las proteínas APOE y Reelina involucradas en la protección de estos individuos funcionan como ligandos de receptores celulares comunes y los autores apuntan que esto podría sugerir un mecanismo común para la resistencia al Alzheimer.
¿Cómo se podría saber si uno tiene estos genes del alzhéimer?
Con estos descubrimientos cabe especular si habrá más personas que cumplan estos rasgos y si habría posibilidad de plantear cribado para encontrarlas. «Mientras que sí resulta sencillo realmente llevar a cabo los estudios genéticos para buscar la variante, sin embargo, estamos ante una mutación muy rara. Por lo que lo más seguro es que se encontrará en pocos pacientes», lamenta Arboleda-Velasquez.
Pese a ello, el investigador de Harvard subraya la participación de la comunidad estudiada. «Es importante reconocer la contribución de los pacientes que con mucho sacrificio participan de los estudios clínicos y genéticos. Ellos son los héroes de este descubrimiento».
Una puerta a nuevas terapias contra el alzhéimer
Si bien en los últimos tiempos llegan buenas noticias en el tratamiento del alzhéimer, la caracterización de estas nuevas pistas genéticas puede suponer opciones a corto plazo. «Cada uno de estos pacientes abre la puerta a varias terapias. Estamos avanzando en el anticuerpo inspirado en el caso de Christchurch y también tenemos progresos en los abordajes inspiradas en el caso de Reelin», detalla Arboleda-Velasquez.
En la actualidad, en EEUU ya está disponible Lecanemab para los pacientes. Se trata de un anticuerpo monoclonal desarrollado por la compañía japonesa Eisai y la estadounidense Biogen indicado para los pacientes que están en las etapas más tempranas de la enfermedad. El fármaco ha logrado reducir la tasa de deterioro cognitivo temprano en un 27% en comparación con un placebo.
Mayo 15/2023 (Diario Médico) – Tomado de Neurología – Estudio en ‘Nature’ Copyright Junio 2018 Unidad Editorial Revistas, S.L.U. Todos los derechos reservados.
may
16

Investigadores de la Universidad de Kanazawa informan en Nano Letters de cómo la microscopía de fuerza atómica de alta velocidad permite comprender procesos relacionados con la enfermedad de Alzheimer. Además, la técnica se revela como una herramienta excelente para estudiar el efecto de fármacos contra la enfermedad.
Según la hipótesis amiloide, la enfermedad de Alzheimer -el tipo más común de demencia- está causada por fallos en la producción, acumulación y eliminación de beta-amiloide (Aβ) en el cerebro. Aβ hace referencia a un grupo de péptidos (fragmentos de proteínas) que con el tiempo forman placas en el cerebro de las personas con enfermedad de Alzheimer. Se han desarrollado fármacos para reducir la agregación de Aβ, pero descubrimientos recientes demuestran que los distintos tipos de agregados de Aβ contribuyen de forma diferente al desarrollo de la enfermedad de Alzheimer. En particular, los agregados intermedios, como las protofibrillas, son más tóxicos que las fibrillas finales propiamente dichas, el principal componente de las placas de Aβ. Por tanto, es necesario comprender con precisión las complejas vías de agregación para seguir desarrollando fármacos eficaces contra la enfermedad de Alzheimer. Kenjiro Ono, de la Universidad de Kanazawa, y sus colegas han logrado ahora visualizar la dinámica estructural de las protofibrillas, así como el efecto de un fármaco desarrollado recientemente a base de anticuerpos anti-Aβ.
Los científicos observaron la formación y la estructura de las protofibrillas de Aβ mediante microscopía de fuerza atómica de alta velocidad (HS-AFM). Este último método se ha revelado en los últimos años como una potente herramienta de nanoimagen para estudiar biomoléculas y su dinámica con alta resolución espaciotemporal. Las observaciones de HS-AFM mostraron que las protofibrillas tienen una estructura nodal, con características estructurales estables -en concreto, el ángulo de unión entre nodos- en varias muestras. Es importante destacar que esta estructura nodal es distinta de las fibrillas maduras propiamente dichas, que tienen una estructura helicoidal.
A continuación, Ono y sus colegas investigaron la disociación de las protofibrillas. Descubrieron que la longitud de las protofibrillas depende de su concentración, lo que sugiere que los agregados pueden disociarse espontáneamente.
Para obtener una visión detallada del funcionamiento de los fármacos anticuerpos anti-Aβ, los investigadores examinaron la unión entre las protofibrillas de Aβ y un nuevo fármaco conocido como lecanemab. Descubrieron que la capacidad de unión (afinidad) de lecanemab por las protofibrillas es casi independiente del tamaño de las protofibrillas; en otras palabras, la afinidad no varía sustancialmente a lo largo del proceso de agregación. Las observaciones de HS-AFM revelaron además que el lecanemab cubre la superficie de pequeños agregados de preprotofibrillas. Al hacerlo, el fármaco inhibe la agregación posterior en protofibrillas, lo que a su vez impide la formación de fibrillas y placas de Aβ propiamente dichas.
Los resultados de Ono y sus colegas aportan pruebas directas de un mecanismo a través del cual un fármaco anticuerpo interfiere en el proceso de agregación de Aβ. En términos más generales, el trabajo confirma la versatilidad del método HS-AFM para estudiar vías bioquímicas. Citas de los científicos: «La HS-AFM de molécula única es una herramienta eficaz para revelar la dinámica estructural de los intermediarios de agregación amiloide transitorios y metaestables y los efectos de los fármacos antiagregantes sobre ellos.»
Mayo 15/2023 (Asia Research News) – Tomado de News Room Copyright 2004 – 2023 Asia Research News
may
13
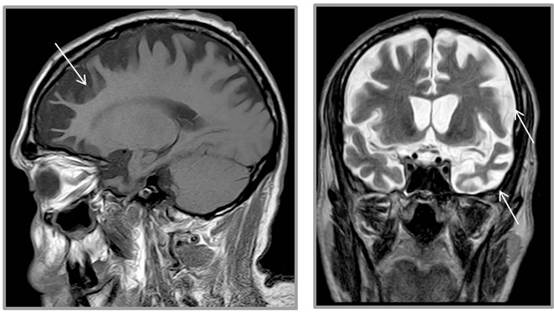
La tomografía por emisión de positrones (PET) de amiloide, muy usada en investigación, tiene también utilidad diagnóstica en pacientes con pérdida de memoria, algo que habían apuntado ya estudios previos pero que ahora se ha confirmado con un ensayo clínico controlado y aleatorizado llamado Estudio de Diagnóstico y Gestión del Paciente de Amypad (Amypad-DPMS), en el que, por parte española, ha participado el Barcelonaβeta Brain Research Center (BBRC), centro de investigación de la Fundación Pasqual Maragall.
Según ha informado hoy el BBRC, el estudio muestra un diagnóstico de alta certeza en el 40% de los pacientes dentro de los tres meses de la visita inicial a la clínica. «Esto corresponde a un porcentaje 3,5 veces superior a pacientes en los que no se habían realizado la PET de amiloide. Además, la PET de amiloide cambió el diagnóstico inicial en el 44% de los casos, frente a solo el 11% en el grupo sin esta prueba».
Para todas las fases de la enfermedad
El estudio, publicado en JAMA Neurology, muestra además que la utilidad diagnóstica de la PET de amiloide se observó «consistentemente» en pacientes beneficiados por la evaluación de biomarcadores (con un deterioro cognitivo leve) y también en los que estaban en una etapa temprana de disfunción clínica, que experimentaban solo quejas cognitivas subjetivas, y en los que ya se hallaban en una etapa tardía de la enfermedad, con demencia.
«Este nuevo ensayo clínico proporciona evidencias sólidas que respaldan la implementación temprana de esta prueba, ya que su uso está asociado a diagnósticos certeros», según Giovanni B. Frisoni, director del Centro de la Memoria del Hospital Universitario de Ginebra e investigador principal del estudio.
«Un diagnóstico seguro y de confianza es fundamental para la eficacia de las terapias modificadoras de la enfermedad, especialmente fármacos antiamiloide, cuya efectividad podría disminuir con el avance de la enfermedad», añade.
Juan Domingo Gispert, responsable del Grupo de Investigación en Neuroimagen del BBRC, que ha liderado la participación del centro en Amypad, ha explicado a este diario, por su parte, que el 40% es «bajo» -«en realidad se buscaba un nivel de certeza muy alto, del 90%»- pero es “reproducible” y está en línea con la dificultad de diagnóstico etiológico que de por sí tienen en enfermedades como el Alzheimer.
Lo que la PET de amiloide permite ver es la presencia o no de placas de la proteína beta amiloide en el cerebro. Si las hay, la prueba es positiva, y si no las hay, negativa. «Esto funciona muy bien en el entorno clínico», asegura el experto.
El resultado de la PET de amiloide es especialmente relevante ante personas con demencia atípica como pueden ser los jóvenes, indica Domingo. También dice que esta tecnología ofrece resultados similares a los de la punción lumbar para el diagnóstico del Alzheimer, que es algo que ya destacó un metaanálisis de 2018. No obstante, destaca que la PET de amiloide resultaría más cara que la punción lumbar y estaría menos accesible para todos los pacientes candidatos a un estudio de imagen con ella.
Para este estudio participaron 844 pacientes de ocho unidades o clínicas de memoria europeos. «Se trata de un estudio prospectivo, multicéntrico, aleatorizado y controlado, y es el más grande de Europa para evaluar el impacto clínico de esta herramienta de diagnóstico en participantes de clínicas de memoria», subraya Daniele Altomare, investigador postdoctoral senior y coordinador del estudio en el Laboratorio de Neuroimagen del Envejecimiento en la Universidad de Ginebra y primer autor del trabajo.
Entre los pacientes reclutados, 794 realizaron la visita a los tres meses y, por lo tanto, fueron considerados para el análisis del resultado principal; algunos experimentaban deterioro cognitivo subjetivo y mayor riesgo de enfermedad de Alzheimer preclínica (239), mientras que otros mostraban deterioro cognitivo leve (318) o demencia (237).
Mayo 12/2023 (Diario Médico) – Tomado de Radiodiagnóstico – Diagnóstico por la imagen Copyright Junio 2018 Unidad Editorial Revistas, S.L.U.
may
10
Una nueva investigación ha descubierto que algunos pacientes con enfermedad de la motoneurona (EMN) y demencia frontotemporal (DFT) son portadores de los mismos defectos genéticos raros que causan otras enfermedades neurodegenerativas.
Investigadores del Centro de Investigación de la MND de la Universidad Macquarie y del Instituto de Investigación Médica Walter y Eliza Hall han identificado los defectos en los genomas de algunas personas con MND y FTD no hereditarias o esporádicas.
La EMN provoca la muerte de las neuronas, o nervios motores, que conectan el cerebro y la médula espinal con los músculos. Son las células que controlan nuestra capacidad para movernos, respirar y tragar. La enfermedad es progresiva y finalmente mortal.
La FTD también causa la muerte de neuronas en parte del cerebro, lo que provoca una serie de síntomas progresivos como pérdida de memoria, comportamiento inusual, cambios de personalidad y problemas de comunicación. Es la misma forma de demencia que se le diagnosticó recientemente al actor Bruce Willis y, a diferencia de la demencia de inicio más antiguo, suele afectar a personas menores de 65 años.
La mayoría de los casos de ambas enfermedades –alrededor del 90% en el caso de la MND y del 60%-70% en la FTD– son esporádicos, y el resto se dan en familias.
Estos defectos genéticos, conocidos como expansiones cortas de repeticiones en tándem, son la causa de más de 20 enfermedades neurodegenerativas, entre ellas las ataxias espinocerebelosas y la distrofia miotónica. Este estudio australiano ha sido la evaluación más completa de estos defectos genéticos en pacientes con EMN y FTD de todo el mundo.
La Dra. Lyndal Henden, investigadora postdoctoral de la Universidad Macquarie, afirma que los resultados fueron una sorpresa.
«Descubrimos que casi el 18% de los pacientes esporádicos de EMN y FTD eran portadores de una expansión repetida del ADN que se cree que está implicada en otras enfermedades degenerativas», afirma.
«Descubrir esta conexión genética entre la EMN y la FTD ofrece una nueva oportunidad de descubrir factores de riesgo comunes para la muerte neuronal, y tendrá implicaciones para comprender ambas enfermedades».
La profesora asociada de la Universidad Macquarie Kelly Williams dirigió el estudio y afirma que el equipo sospechaba que podría haber cierto solapamiento con otras enfermedades, pero no hasta tal punto.
«Esto sugiere factores de riesgo compartidos entre estas enfermedades, mecanismos compartidos que provocan la muerte de los nervios, y quizá estrategias terapéuticas compartidas en el futuro», afirma.
«Aunque las causas de la EMN esporádica y la FTD siguen siendo desconocidas, éste es un paso importante en un esfuerzo a largo plazo para identificar los factores de riesgo de desarrollar una de estas enfermedades».
Ahora se puede empezar a trabajar para entender cómo estas expansiones repetidas compartidas contribuyen a la muerte neuronal.
El estudio, publicado en el último número de la revista Science Advances, es la culminación de 10 años de investigación que no habrían sido posibles sin la colaboración de pacientes con EMN y FTD, que han donado muestras biológicas para ADN tanto en la Universidad Macquarie como en la Universidad de Sídney.
Mayo 10/2023 (MedicalXpress) – Tomado de Genetics – Neuroscience Copyright Medical Xpress 2011 – 2023 powered by Science X Network.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
may
9
Científicos de Singapur han demostrado el papel fundamental que desempeña una proteína transportadora especial en la regulación de las células cerebrales que garantizan la protección de los nervios mediante cubiertas llamadas vainas de mielina. Los hallazgos, publicados por investigadores de la Facultad de Medicina Duke-NUS y la Universidad Nacional de Singapur en la revista Journal of Clinical Investigation, podrían ayudar a reducir los efectos nocivos del envejecimiento en el cerebro.
Las vainas de mielina, una membrana aislante que recubre los nervios, facilitan la conducción rápida y eficaz de las señales eléctricas por todo el sistema nervioso. Cuando la vaina de mielina se daña, los nervios pueden perder su capacidad de funcionamiento y provocar trastornos neurológicos. Con el envejecimiento, las vainas de mielina pueden empezar a degenerar de forma natural, lo que suele ser la causa de que los ancianos pierdan sus capacidades físicas y mentales.
«La pérdida de vainas de mielina se produce durante el proceso normal de envejecimiento y en enfermedades neurológicas como la esclerosis múltiple y el Alzheimer», explica el Dr. Sengottuvel Vetrivel, investigador principal del Programa de Trastornos Cardiovasculares y Metabólicos (CVMD) de la Universidad Duke-NUS e investigador principal del estudio. «Desarrollar terapias para mejorar la mielinización -la formación de la vaina de mielina- en el envejecimiento y la enfermedad es de gran importancia para aliviar las dificultades causadas por el declive de la mielinización».
Para allanar el camino al desarrollo de tales terapias, los investigadores trataron de comprender el papel de Mfsd2a, una proteína que transporta lisofosfatidilcolina (LPC) -un lípido que contiene un ácido graso omega-3- al cerebro como parte del proceso de mielinización. Por lo que se sabe, los defectos genéticos en el gen Mfsd2a conducen a una mielinización significativamente reducida y a un defecto congénito llamado microcefalia, que hace que la cabeza del bebé sea mucho más pequeña de lo que debería.
En modelos preclínicos, el equipo demostró que la eliminación de Mfsd2a de las células precursoras que maduran hasta convertirse en células productoras de mielina -conocidas como oligodendrocitos- en el cerebro provocaba una mielinización deficiente tras el nacimiento. Otras investigaciones, incluida la secuenciación de ARN unicelular, demostraron que la ausencia de Mfsd2a provocaba la reducción del conjunto de moléculas de ácidos grasos -en particular de grasas omega-3- en las células precursoras, lo que impedía que estas células maduran hasta convertirse en oligodendrocitos productores de mielina.
«Nuestro estudio indica que los lípidos omega-3 LPC actúan como factores en el cerebro que dirigen el desarrollo de los oligodendrocitos, un proceso crítico para la mielinización cerebral», explicó el profesor David Silver, autor principal del estudio y Director Adjunto del Programa CVMD. «Esto abre posibles vías para desarrollar terapias y suplementos dietéticos basados en lípidos omega-3 LPC que podrían ayudar a retener la mielina en el cerebro que envejece, y posiblemente para tratar a pacientes con trastornos neurológicos derivados de una mielinización reducida».
Anteriormente, el profesor Silver y su laboratorio descubrieron el Mfsd2a y colaboraron estrechamente con otros equipos para determinar la función de los lípidos LPC en el cerebro y otros órganos. La investigación actual aporta más datos sobre la importancia del transporte de lípidos para el desarrollo de células precursoras de oligodendrocitos.
«Ahora nos proponemos realizar estudios preclínicos para determinar si los omega-3 LPC dietéticos pueden ayudar a remielinizar los axones dañados en el cerebro», añadió el profesor Silver. «Nuestra esperanza es que los suplementos que contienen estas grasas puedan ayudar a mantener -o incluso mejorar- la mielinización cerebral y la función cognitiva durante el envejecimiento».
Mayo 8/2023 (Asia Research News) – Tomado de News Room Copyright 2004 – 2023 Asia Research News
may
8

Investigadores de la Universidad de Minnesota en las Ciudades Gemelas han desarrollado una innovadora técnica de diagnóstico que permitirá una detección más rápida y precisa de las enfermedades neurodegenerativas. El método abrirá probablemente una puerta al tratamiento precoz y la mitigación de diversas enfermedades que afectan a los seres humanos, como el Alzheimer y el Parkinson, y otras similares que afectan a los animales, como la caquexia crónica.
Su nuevo estudio se publica en Nano Letters, una revista de primera línea en el campo de la nanotecnología editada por la American Chemical Society.
«Este trabajo se centra principalmente en la caquexia crónica del ciervo, pero nuestro objetivo final es ampliar la tecnología a un amplio espectro de enfermedades neurodegenerativas, siendo el Alzheimer y el Parkinson los dos objetivos principales», afirma Sang-Hyun Oh, coautor principal del trabajo y catedrático McKnight del Departamento de Ingeniería Eléctrica e Informática de la Universidad de Minnesota. «Nuestra visión es desarrollar técnicas de diagnóstico ultrasensibles y potentes para diversas enfermedades neurodegenerativas, de modo que podamos detectar biomarcadores en una fase temprana, lo que quizá permita disponer de más tiempo para el despliegue de agentes terapéuticos que puedan ralentizar la progresión de la enfermedad. Queremos ayudar a mejorar la vida de millones de personas afectadas por enfermedades neurodegenerativas».
Enfermedades neurodegenerativas como el Alzheimer, el Parkinson, la enfermedad de las vacas locas y la caquexia crónica (ampliamente extendida entre los ciervos) comparten una característica común: la acumulación de proteínas mal plegadas en el sistema nervioso central. Detectar estas proteínas mal plegadas es crucial para comprender y diagnosticar estos trastornos devastadores. Sin embargo, los métodos de diagnóstico existentes, como el ensayo inmunoenzimático y la inmunohistoquímica, pueden ser caros, lentos y limitados en cuanto a la especificidad de los anticuerpos.
El método de los investigadores de la Universidad de Minnesota, denominado Nano-QuIC (Nanoparticle-enhanced Quaking-Induced Conversion), mejora significativamente el rendimiento de los métodos avanzados de detección del mal plegamiento de proteínas, como el ensayo RT-QuIC (Real-Time Quaking-Induced Conversion) de los NIH Rocky Mountain Laboratories.
El método RT-QuIC consiste en agitar una mezcla de proteínas normales con una pequeña cantidad de proteína mal plegada, lo que desencadena una reacción en cadena que hace que las proteínas se multipliquen y permite la detección de estas proteínas irregulares. Utilizando muestras de tejido de ciervos, el equipo de la Universidad de Minnesota demostró que la adición de nanopartículas de sílice de 50 nanómetros a los experimentos RT-QuIC reduce drásticamente los tiempos de detección de unas 14 horas a sólo cuatro y multiplica por 10 la sensibilidad.
Un ciclo de detección típico de 14 horas significa que un técnico de laboratorio sólo puede realizar una prueba por jornada laboral normal. Sin embargo, con un tiempo de detección inferior a cuatro horas, los investigadores pueden realizar tres o incluso cuatro pruebas al día.
Disponer de un método de detección más rápido y preciso es especialmente importante para comprender y controlar la transmisión de la caquexia crónica, una enfermedad que se está extendiendo entre los ciervos de Norteamérica, Escandinavia y Corea del Sur. Los investigadores creen que el Nano-QuIC podría llegar a ser útil para detectar enfermedades de plegamiento erróneo de proteínas en humanos, en concreto el Parkinson, la enfermedad de Creutzfeldt-Jakob, el Alzheimer y la ELA.
«Las pruebas para detectar estas enfermedades neurodegenerativas tanto en animales como en seres humanos han supuesto un gran reto para nuestra sociedad», afirma Peter Larsen, coautor principal del artículo y profesor adjunto del Departamento de Veterinaria y Ciencias Biomédicas de la Universidad de Minnesota.
«Lo que estamos viendo ahora es este momento realmente emocionante en el que están surgiendo nuevas pruebas de diagnóstico de nueva generación para estas enfermedades. El impacto de nuestra investigación es que mejora en gran medida esas pruebas de nueva generación, las hace más sensibles y más accesibles».
Mayo 8/2023 (EurekaAlert!) – Tomado de News Releases Copyright 2023 by the American Association for the Advancement of Science (AAAS)
Noticias anteriores a enero de 2010
Suscripción AL Día
