
abr
21
 La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
De acuerdo con la literatura médica, es considerada poco frecuente y que aqueja a hombres y mujeres de cualquier edad, sin importar el origen étnico, e incluso puede originarse en cualquier etapa de vida del paciente desde lactantes hasta adultos.
Es una enfermedad neuromuscular hereditaria autosómica recesiva y de origen genético, causada por un gen que genera el déficit de la enzima alfa-glucosidasa ácida o maltasa ácida.
Ello permite la acumulación de una sustancia en los músculos denominada glucógeno, ocasionando pérdida de masa muscular, así como debilidad muscular en las extremidades inferiores.
Igualmente puede afectar los músculos del tronco, brazos y hombros, y la gravedad y síntomas varían en cada paciente.
Entre los malestares relacionados con esta dolencia genética están la dificultad para levantarse, al estar sentado, para caminar, subir escaleras, masticar y la debilidad muscular progresiva.
Una persona aquejada por la Enfermedad de Pompe, también puede presentar reflujo gastroesofágico, disnea, caídas frecuentes, agitación tras realizar esfuerzo físico, dolor de cabeza por las mañanas y somnolencia durante el día debido a la ortopnea.
El nombre de esta dolencia es en honor al patólogo holandés Joannes Pompe, quien presentó el primer caso en el año 1930, y cada 15 de abril es celebrado el Día Internacional de la Enfermedad de Pompe.
15 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
mar
25
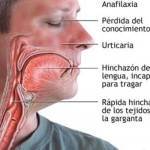 Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
El trabajo, publicado en la revista científica Frontiers in Immunology, destaca la identificación de varios casos portadores de la misma variante genética (T328K gen F12), todos ellos concebidos mediante reproducción asistida a través de un único donante.
Angioedema hereditario
El angioedema hereditario es una enfermedad genética muy poco frecuente que provoca hinchazón en la piel, aparato digestivo o vías respiratorias; unos síntomas que afectan al 60-80 % de las mujeres portadoras, frente a una probabilidad muy baja en los hombres.
La investigación arrancó con una paciente que presentaba episodios recurrentes de hinchazón en la cara, sin responder a los tratamientos antialérgicos habituales, y cuyo análisis genético identificó la variante T328K.
Esta fue también detectada posteriormente en otras personas concebidas con el esperma del mismo donante, mientras que no estaba presente en la línea materna analizada.
Ante la sospecha de que la mutación genética podía proceder del padre, los investigadores contactaron con la clínica de fertilidad y, tras coordinar un estudio con el banco de esperma, pudieron confirmar que este era portador heterocigoto de la variante.
Notificación de la clínica
La clínica notificó entonces a las mujeres que habían utilizado el esperma de dicho donante, un procedimiento que permitió identificar a más portadores, algunos de ellos sin manifestaciones sintomáticas.
«Desde el punto de vista genético, este caso es especialmente relevante porque demuestra cómo una variante patogénica puede pasar desapercibida en un donante asintomático y transmitirse a varios descendientes», explica el jefe del grupo de Inmunología Traslacional del Vall d’Hebron Institut de Recerca (VHIR), Roger Colobran.
Los investigadores subrayan que este caso pone de relieve los retos que plantean las enfermedades genéticas hereditarias asintomáticas en los programas de donación de gametos, ya que el cribado genético de los donantes se centra principalmente en enfermedades infecciosas.
En este sentido, señalan que, en regiones donde esta variante es más prevalente, podría valorarse el cribado dirigido al gen F12.
20 marzo 2026 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jun
15
 A 10 años de celebrarse en esta fecha el Día Internacional de Sensibilización sobre el Albinismo, continúan los esfuerzos por garantizar los derechos y eliminar la discriminación de personas con este trastorno de salud.
A 10 años de celebrarse en esta fecha el Día Internacional de Sensibilización sobre el Albinismo, continúan los esfuerzos por garantizar los derechos y eliminar la discriminación de personas con este trastorno de salud.
Establecida por resolución de las Asamblea General de las Naciones Unidas (ONU) en 2014, cada 13 de junio se conmemora esta efeméride que pretende crear conciencia mundial, así como evitar el maltrato y discriminación hacia las personas albinas, y acabar con el estigma y falsas creencias sobre esta condición, donde la superstición y el fanatismo no tienen cabida.
En esta jornada, la ONU destacó los incansables esfuerzos de los grupos de albinismo de todo el mundo y, a la par, instó a reflexionar sobre los continuos cambios jurídicos, políticos y prácticos que aún son necesarios para garantizar el disfrute pleno e igualitario de los derechos de las personas con albinismo.
Según los expertos, este es un trastorno poco frecuente, no contagioso, hereditario y congénito.
En casi todos los tipos de albinismo, ambos progenitores deben ser portadores del gen para que se transmita el trastorno, aunque ellos mismos no lo presenten.
Se da en ambos sexos, es independiente del origen étnico y puede existir en cualquier territorio.
Se caracteriza por la ausencia de pigmentación (melanina) en la piel, el cabello y los ojos, lo que causa sensibilidad al sol y a la luz intensa, y provoca que casi todas las personas con albinismo padezcan deficiencias visuales y sean propensas a sufrir cáncer de piel.
Los especialistas estiman que en América del Norte y Europa una de cada 17 000 a 20 000 personas presenta algún tipo de albinismo.
En tanto, la afección es mucho más común en África subsahariana, donde se cree que una de cada 1 400 personas está afectada en Tanzania y hay prevalencias de hasta uno de cada 1000 individuos en ciertas poblaciones de Zimbabwe y otros grupos étnicos concretos en África Meridional.
La falta de melanina en los albinos hace que sean extremadamente vulnerables al cáncer de piel, por lo que en algunos países la mayoría de ellos muere por esta causa entre los 30 y los 40 años de edad.
Si pudieran ejercer plenamente su derecho a la salud, los albinos podrían tratarse de este tipo de cáncer ya que es prevenible en un alto porcentaje —con revisiones médicas regulares, protectores solar, gafas de sol y ropa de protección solar— sin embargo, en muchas naciones no tienen acceso a estos recursos ya que se encuentran fuera del sistema.
13 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
jun
7
 El Premio Princesa de Asturias de Investigación Científica fue anunciado hoy en Oviedo, donde el jurado galardonó a expertos que desarrollaron fármacos contra la diabetes y la obesidad.
El Premio Princesa de Asturias de Investigación Científica fue anunciado hoy en Oviedo, donde el jurado galardonó a expertos que desarrollaron fármacos contra la diabetes y la obesidad.
Los especialistas proceden de Estados Unidos, Canadá y Dinamarca, considerados referentes mundiales en el campo de la endocrinología. Son ellos, Daniel J. Drucker, Jeffrey M. Friedman, Joel F. Habener, Jens Juul Holst y Svetlana Mojsov.
«Las investigaciones de los científicos laureados han establecido las bases endocrinas de la diabetes y la obesidad, patologías prominentes que son un problema global de salud pública sin tratamiento efectivo hasta la fecha», destacó el jurado.
En un primer momento, los científicos desarrollaron un tratamiento para la diabetes, el cual posteriormente se aplicó con éxito a la lucha contra la obesidad y el sobrepeso. También, resultó útil frente a patologías asociadas, a la sazón cardiovasculares.
De forma separada, aunque con un nivel de coordinación e intercambio de criterios, Habener y Drucker desde Estados Unidos, y de forma paralela la bioquímica Mojsov, se enfocaron en nuevos tipos de hormonas intestinales relacionadas con la obesidad.
En tanto, en Holst descubrió en la Universidad de Copenhague que los niveles de insulina se disparaban y los niveles de azúcar disminuían en pacientes sometidos a una cirugía intestinal y relacionó esos cambios con varias hormonas.
05 junio 2024|Fuente: Prensa Latina |Tomado de |Noticia
may
2
 Un equipo internacional de investigadores ha descubierto más de cien nuevas regiones del genoma humano (o loci), que parecen influir en la tensión arterial de una persona.
Un equipo internacional de investigadores ha descubierto más de cien nuevas regiones del genoma humano (o loci), que parecen influir en la tensión arterial de una persona.
Además, varios de ellos pueden ser relevantes para el metabolismo del hierro y un tipo de receptor celular conocido como receptores adrenérgicos.
El estudio, liderado por los Institutos Nacionales de la Salud estadounidenses (NIH) y el Instituto Nacional de Investigación del Genoma Humano (NHGRI) y publicado este martes en la revista Nature Genetics, es uno de los mayores estudios genómicos sobre la presión arterial realizados hasta la fecha, con datos de más de un millón de participantes.
Los resultados servirán para comprender mejor cómo se regula la presión arterial y encontrar posibles nuevas dianas farmacológicas.
«Nuestro estudio ayuda a explicar una proporción mucho mayor de las diferencias entre la presión arterial de dos personas de lo que se sabía hasta ahora», dice Jacob Keaton, científico del NHGRI y primer autor del estudio.
Además, «conocer el riesgo de una persona de desarrollar hipertensión podría conducir a tratamientos a medida, que tienen más probabilidades de ser eficaces», puntualiza.
Para comprender la genética de la presión arterial, el equipo combinó cuatro grandes conjuntos de datos procedentes de estudios de asociación de todo el genoma de la presión arterial y la hipertensión.
Tras analizar los datos, hallaron más de 2 000 loci genómicos relacionados con la presión arterial, incluidas 113 regiones nuevas.
Entre los loci genómicos recién descubiertos, varios residen en genes que desempeñan un papel en el metabolismo del hierro, lo que confirma estudios anteriores según los cuales los niveles elevados de hierro acumulado pueden contribuir a las enfermedades cardiovasculares.
El equipo también confirmó la asociación entre las variantes del gen ADRA1A y la presión arterial.
ADRA1A codifica un tipo de receptor celular, denominado receptor adrenérgico, que actualmente es diana de la medicación contra la tensión arterial, lo que sugiere que otras variantes genómicas descubiertas en el estudio también pueden tener el potencial de ser dianas farmacológicas para alterar la tensión arterial.
«Este estudio demuestra que estos grandes estudios de asociación de todo el genoma tienen relevancia clínica para encontrar nuevas dianas farmacológicas y son necesarios para descubrir más dianas farmacológicas a medida que avanzamos», defiende Keaton.
A partir de estos análisis, los investigadores pudieron calcular una puntuación de riesgo poligénico, que combina los efectos de todas las variantes genómicas para predecir la presión arterial y el riesgo de hipertensión.
Estas puntuaciones de riesgo poligénico pueden ser una herramienta útil en la medicina de precisión, pero se necesitan datos genómicos más diversos para que puedan aplicarse ampliamente en la atención sanitaria rutinaria.
Aunque los datos recopilados procedían en su mayoría de personas de ascendencia europea, los investigadores descubrieron que las puntuaciones de riesgo poligénico también eran aplicables a personas de ascendencia africana.
Casi la mitad de los adultos de Estados Unidos padecen presión arterial alta o hipertensión, una dolencia que suele ser hereditaria, lo que significa que, junto al estilo de vida (sendentarismo, tabaquismo o estrés) y la dieta, hay un componente genético en el desarrollo de esta afección.
Cuando la tensión arterial es demasiado alta de forma constante, puede dañar el corazón y los vasos sanguíneos del cuerpo y provocar cardiopatías, enfermedades renales, accidentes cerebrovasculares y otras afecciones.
30 abril 2024|Fuente: EFE |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2023. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
abr
24
 Actualmente existen alrededor de 6 313 enfermedades raras identificadas, aunque esta cifra es variable ya que, según ha asegurado el miembro de la junta directiva de la Federación Española de Enfermedades Raras (FEDER), Daniel de Vicente, «todavía hay más de 7 000 enfermedades en el mundo que pueden ser catalogadas como poco frecuentes», pero, de todas ellas, solo el 6 % por ciento disponen de un tratamiento eficaz.
Actualmente existen alrededor de 6 313 enfermedades raras identificadas, aunque esta cifra es variable ya que, según ha asegurado el miembro de la junta directiva de la Federación Española de Enfermedades Raras (FEDER), Daniel de Vicente, «todavía hay más de 7 000 enfermedades en el mundo que pueden ser catalogadas como poco frecuentes», pero, de todas ellas, solo el 6 % por ciento disponen de un tratamiento eficaz.
«Todas las enfermedades raras carecen muchas veces de un conocimiento estricto por parte de los profesionales y hay una falta de investigación en todas ellas. Esto conlleva que el número de terapias sea muy reducido y que solo el 6 % de estas enfermedades tengan un tratamiento eficaz. Esto tiene un gran impacto en la calidad de vida de los pacientes y sus familias ya que muchas son degenerativas», ha explicado Daniel de Vicente durante una jornada sobre la situación actual de las enfermedades raras, organizada por la Fundación Ramón Areces con el Centro
de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) y FEDER.
Durante la jornada los expertos han coincidido en que en España existe una falta de acceso y equidad a las terapias y tratamientos existentes ya que, de los 147 medicamentos huérfanos autorizados en Europa, solo 78 están financiados en España y el tiempo medio de acceso a las terapias es de 23 meses, según ha aseverado Daniel de Vicente recalcando que es «una situación compleja».
En este sentido, el presidente de FEDER, Juan Carrión, ha incidido en la necesidad de seguir investigando las enfermedades raras para poder diagnosticarlas y tratarlas a tiempo ya que, «de las más de 6 000 enfermedades raras identificadas, solo el 30 % de estas enfermedades se investigan, se invierte el 1,43 % del Producto Interno Bruto (PIB) en investigación de enfermedades raras, un 1,28 % por debajo de otros países».
«Hay que garantizar acceso a los tratamientos ya que esto no es una realidad porque supeditamos el acceso al código postal de residencia del paciente ya que, hasta en una misma comunidad autónoma, hay diferencias entre los hospitales que disponen o no del medicamento. Hay una necesidad de abordar un Plan de Acción a nivel estatal», ha afirmado Carrión señalando que, al día de hoy, «solo hay siete CCAA que disponen de un Plan de Atención Integral o estrategias» y que se necesita «presupuesto para que estos planes se lleven a cabo».
INTELIGENCIA ARTIFICIAL Y GENÓMICA DE PRECISIÓN: LA CLAVE DEL FUTURO
Una de las claves del futuro del diagnóstico y tratamiento de las enfermedades raras reside en la genómica de precisión ya que el 80 % de estas enfermedades tienen un origen genético. Por ello, los expertos han insistido en la necesidad de que España disponga de una especialidad de genética, que a día de hoy no existe en el SNS, a diferencia del resto de países de Europa que sí que tienen la especialidad de genética.
La aplicación de la Inteligencia Artificial supone un gran reto en el diagnóstico y tratamiento de las enfermedades raras en muchos aspectos ya que, por ejemplo, «tiene un gran potencial en la clasificación de las enfermedades raras», un aspecto crucial que a día de hoy no se consigue porque «si no se sabe cuantas enfermedades hay, es imposible clasificarlas», aunque el uso de la IA siempre debe acompañarse ‘de la supervisión de un especialista», tal y como ha explicado el miembro del Instituto de Genética Médica y Molecular, Hospital La Paz, Julián Nevado.
«La IA ayudará para automatizar la resolución de problemas, mejorar la capacidad de tomar decisiones, caracterizar de forma más precisa los estados de salud, incluyendo las EERR. Necesitamos mejorar la precisión en diagnóstico pero también la clasificación de las enfermedades. Siempre bajo la supervisión de los profesionales sanitarios», ha indicado Julián Nevado.
Por su parte, la biología molecular y el uso de las tecnologías genómicas también son esenciales y pueden tener muchas ventajas en las enfermedades raras ya que permiten agilizar el diagnóstico e implementar tratamientos más personalizados.
En este sentido, los expertos han apuntado a la metagenómica, cuya base principal está en la biología molecular, y que pretende no centrarse en el organismo individual, sino en la microbiología, en el ADN y ARN presente en su hábitat natural como el intestino humano.
El uso de la metagenómica en enfermedades raras permite identificar microorganismos presentes en muestras clínicas, como heces o saliva, que podrían estar relacionados con la enfermedad poco frecuente; estudiar el microbioma intestinal y su posible influencia en la enfermedad poco frecuente; ayudar en la identificación de biomarcadores microbianos o genéticos asociados con estas EERR, que podrían usarse para el diagnóstico temprano, la estratificación de pacientes o el monitoreo de la progresión de la enfermedad.
«Con la genómica podemos abarcar muchas de las enfermedades genéticas. Tendríamos que estar más centrados en los procesos y generar una cartera de diagnósticos que nos ayude en los pacientes. Es un proceso complejo, hay que tener investigaciones y validación sólidas y hay que integrar nuevos perfiles de profesionales ya que para la genómica necesitamos bioinformáticos para interpretar y analizadores de datos», ha
señalado Nevado
Entre los avances terapéuticos también destacan los avances bioquímicos para el diagnóstico de las EERR que incluyen: la secuenciación masiva del estudio del genoma, que ha tenido un desarrollo exponencial en los últimos años siendo más eficiente, rápida y barata; las terapias CRISPR-CAS, que consisten en se trata de una tecnología de «edición genética» con enorme potencial, inicialmente como herramienta terapéutica; y, por último, los marcadores bioquímicos, que permiten monitorizar las respuestas a los tratamientos y agilizar el diagnóstico.
Asimismo, destacan los avances en los diagnósticos por imagen que, aunque en los últimos años «no ha salido una tecnología mejor que la resonancia magnética», esta técnica «ha mejorado mucho en los últimos años» y «sigue siendo la reina», según ha asegurado la neuropediatra del Hospital Universitario Central de Asturias, Raquel Blanco.
Así, la doctora Blanco ha detallado que las técnicas de RM ahora son «mucho más finas y con mayor resolución» ya que cada vez se obtienen mejores imágenes en un menor tiempo. Entre las técnicas destaca la resonancia PET-TC que permite el diagnóstico de algunas enfermedades raras como la encefalitis en un menor tiempo posible.
PROGRAMA DE PATOLOGÍAS POCO FRECUENTES
Durante el encuentro, la Fundación Ramón Areces ha anunciado un nuevo programa específico orientado a las patologías poco frecuentes en el que financiará un proyecto de investigación transversal y multidisciplinario que incluirá una dotación específica para laboratorios en enfermedades raras. También se dotará económicamente de 10 o 12 becas predoctorales para aquellos estudiantes que, habiendo acabado la
carrera, quieran realizar una tesis doctoral en este campo, según ha explicado el presidente del Consejo Científico de la Fundación, Emilio Bouza.
Dentro de ese compromiso con las patologías poco frecuentes, también se celebrará todos los años una jornada como la de este martes para poder conocer la evolución de la investigación en estas dolencias.
23 abril 2024|Fuente: Europa Press |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2023. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
