
may
13
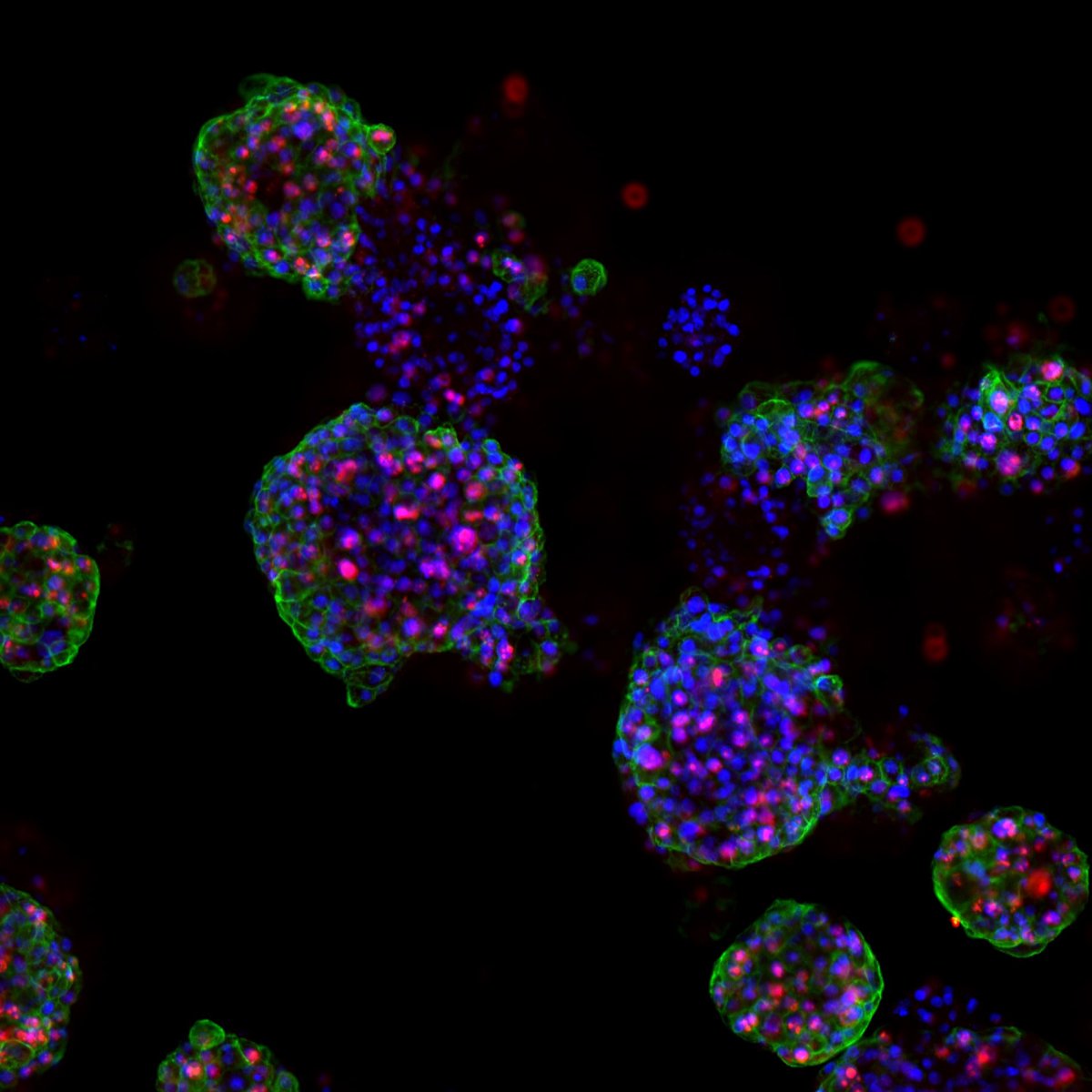
Cáncer de cabeza y cuello (CCC) es un término genérico que engloba varios tipos de cáncer, entre ellos el más frecuente, el carcinoma de células escamosas de cabeza y cuello (CCECC). Los pacientes con CCC pueden ser tratados con una combinación de cirugía, radioterapia y/o quimioterapia. Sin embargo, el tratamiento no siempre es eficaz.
Rosemary Millen, primera autora compartida de un artículo publicado recientemente en la revista Med, explica: «Estos tratamientos provocan efectos secundarios graves, por lo que algunos pacientes no pueden terminarlos. Inclusive después de someterse a un tratamiento tan duro, el 60% de los pacientes recae». La limitada eficacia de los tratamientos podría explicarse en parte por el hecho de que la composición genética del tumor difiere mucho de un paciente a otro. En consecuencia, la terapia más adecuada es diferente para cada paciente.
«Los clínicos disponen actualmente de herramientas limitadas para distinguir adecuadamente este hecho. Por tanto, existe una necesidad urgente de mejores biomarcadores: indicadores que podamos medir en cada paciente individual para determinar de qué tratamiento se beneficiaría más. En el mejor de los casos, este enfoque personalizado evitaría sobrecargar innecesariamente a los pacientes con tratamientos que podrían no funcionar, y conduciría a un mejor pronóstico», explica Else Driehuis, autora correspondiente del artículo.
Creación de un biobanco
Un primer paso hacia esos tratamientos personalizados es desarrollar mejores modelos que reflejen con mayor precisión la variabilidad del tumor. Por ello, los investigadores crearon un biobanco de organoides de CCC: versiones en miniatura de tumores de pacientes cultivados en el laboratorio. Millen afirma: «En última instancia, el objetivo de este biobanco sería utilizar los organoides para guiar las decisiones de tratamiento personalizado en la clínica. Por supuesto, antes de poder hacerlo, primero necesitamos investigar si la respuesta al tratamiento en los organoides se correlaciona con la respuesta observada en los pacientes.»
El equipo pudo cultivar organoides de CCC a partir de los tejidos de los pacientes y confirmó que estos «mini tumores» se parecían mucho a los tumores de los pacientes, ya que conservaban las mismas características histológicas y genéticas. Millen afirma: «A continuación, tratamos los organoides con varios tipos de terapia y medimos la eficacia del tratamiento determinando cuántas células de los organoides morían. El siguiente paso fue correlacionar esto con la respuesta al tratamiento en los pacientes».
Comparación de organoides y pacientes
Tras tratar los organoides con radioterapia, el equipo demostró que la respuesta de los organoides se asemejaba a la de los pacientes. «Por tanto, los organoides tienen potencial para predecir los resultados en los pacientes. La correlación entre la respuesta de los organoides y la de los pacientes existía en los casos en que éstos recibían radioterapia adyuvante, es decir, se utilizaba radiación además de la resección quirúrgica del tumor. En los casos en que los pacientes recibieron radioterapia como tratamiento primario, tenemos que investigarlo más a fondo», explica Millen.
Los investigadores también estudiaron el efecto de la quimiorradioterapia, una combinación de radiación y quimioterapia. «Aquí demostramos que dos fármacos quimioterápicos específicos, el cisplatino y el carboplatino, tienen un efecto radiosensibilizador en los organoides. Esto significa que hacen que las células tumorales sean más sensibles a la radioterapia. Estos resultados son coherentes con lo que vemos en la clínica y, por tanto, subrayan de nuevo el potencial predictivo de los organoides en este escenario», afirma Driehuis.
Implicaciones clínicas
Algunos de los descubrimientos realizados por el equipo podrían tener implicaciones para los pacientes de CCC en un futuro próximo. Por ejemplo, demostraron que el fármaco cetuximab hacía que los organoides tumorales fueran menos sensibles a la radioterapia.
Driehuis explica: «Esto es sorprendente, porque la combinación de este fármaco y la radioterapia se administra a algunos pacientes de CCC en la clínica hoy en día. En los pacientes es difícil distinguir las contribuciones individuales del fármaco y la radioterapia al efecto global de este tratamiento combinado, pero en los organoides podemos separarlas».
«Nuestros resultados encajan con datos publicados recientemente que muestran que la supervivencia de los pacientes tratados con cetuximab y radioterapia es peor en comparación con el tratamiento con radioterapia sola. La implicación clínica de estos hallazgos sería que es mejor esperar con cetuximab hasta después de la radioterapia, pero este cambio en el momento del tratamiento es algo que habría que probar en los pacientes, por supuesto.»
El equipo también demostró que un inhibidor de la PRMT5, un nuevo fármaco que ya se está probando en ensayos clínicos para otros tipos de cáncer, podría ser eficaz en un subgrupo de pacientes con CCC.
Millen afirma: «Secuenciamos el ADN de los organoides para investigar la relación entre mutaciones genéticas específicas y la respuesta a los tratamientos. Al hacerlo, descubrimos que los tumores con pérdida del gen CDKN2A respondían al tratamiento con este nuevo fármaco. Sería muy interesante comprobar si este efecto se da también en los pacientes, sobre todo teniendo en cuenta que esta mutación está presente en más del 50% de los casos de CCECC.»
Driehuis concluye: «En conjunto, nuestros resultados ponen de relieve la relevancia clínica de los organoides derivados de tejido tumoral de pacientes. Uno de los próximos pasos sería diseñar un ensayo clínico, para utilizar realmente los organoides para guiar las decisiones de tratamiento en pacientes con CCC.»
Mayo 13/2023 (MedicalXpress) – Tomado de Oncology & Cancer – Biomedical technology Medical Xpress 2011 – 2023 powered by Science X Network.
may
12

El 14 de abril de 2003 es una fecha importante para la historia de la ciencia. Aquel día de hace poco más de 20 años se anunció el fin del Proyecto Genoma Humano: la secuencia esencial de nuestro ADN había conseguido descifrarse después de muchos años de esfuerzo.
No obstante, ese ‘mapa’, que costó 3.000 millones de dólares y que comenzó a usarse como referencia del genoma humano, no estaba completo. Tenía lagunas en varias regiones genéticas y se basaba principalmente en el ADN de unos pocos individuos de origen europeo. Y aunque en estas dos décadas se han producido avances tecnológicos que han permitido ‘cartografiar’ esas lagunas -la secuencia completa se obtuvo en 2022- y abaratar el proceso, seguía faltando una referencia que fuera más global y diversa.
A partir de ahora, gracias a un consorcio internacional financiado por el Instituto de Investigación Nacional del Genoma Humano de EEUU, ese recurso -un pangenoma de referencia- estará disponible.
El nuevo ‘mapa’, que se presenta como un primer borrador, incluye la secuencia genética completa de 47 individuos de diferentes orígenes, lo que proporciona información detallada de 94 genomas debido a que cada individuo lleva en su ADN dos copias de genes ‘heredados’ de su padre y su madre. El objetivo del proyecto es seguir añadiendo datos al ‘mapa’, por lo que prevé que a mediados de 2024 incluya información genética de 350 personas de ascendencia étnica diversa.
«Hasta ahora, la referencia que usaba la comunidad científica estaba incompleta y carecía de diversidad», señaló en rueda de prensa Benedict Paten, director asociado del Instituto de Genómica Santa Cruz de la Universidad de California y uno de los líderes del proyecto. Este nuevo recurso, en cambio, proporciona una imagen más completa y permitirá realizar análisis más certeros a la hora de caracterizar la variabilidad genética de la población humana sea cual sea su origen, destacó.
De hecho, el nuevo pangenoma ya ha sacado a la luz más de 100 millones de nuevas bases -cada una de las letras que componen el genoma-, y ha destapado nuevos alelos en regiones estructuralmente complejas del genoma que hasta ahora no figuraban en el genoma de referencia. Los detalles de la investigación se publican en cuatro artículos en el último número de las revistas Nature y Nature Biotechnology.
Mediante técnicas computacionales de última generación, los investigadores han sido capaces de construir un recurso que, en lugar de ser único y lineal, como era hasta ahora la referencia GRCh38 que se utilizaba, aporta distintas versiones de una misma secuencia al mismo tiempo, lo que proporciona a los investigadores un mayor abanico de opciones para sus análisis. En el proyecto ha participado un equipo del Centro de Supercomputación de Barcelona liderado por Santiago Marco-Sola.
Qué supone un nuevo pangenoma para la investigación
«Hasta ahora nos hemos contentado con una sola secuencia del genoma que en su día se decidió arbitrariamente que era la secuencia referencia, formada por trozos de secuencia de un puñado de personas de ascendencia principalmente europea. Y si bien esto ha tenido una gran utilidad, también tiene muchas limitaciones», señala Jorge Ferrer, investigador del Centro de Regulación Genómica de Barcelona (CRG). «Por ejemplo, aunque resulte sorprendente, a cada uno de nosotros nos pueden faltar o sobrar unos cuantos trozos muy grandes del genoma. Si el pedazo de genoma escogido para ser la referencia es de alguien que no tiene ese trozo (o lo tiene suficientemente alterado), el mapa de referencia que utilizamos actualmente no serviría para una persona que tiene una mutación que afecta esa parte», aclara. Para complicar aún más las cosas, continúa, «el genoma puede variar enormemente en diferentes partes del mundo. Y si el mapa de referencia está hecho con variantes europeas, tiene menor utilidad para interpretar el genoma de una persona de Camerún o China».
El trabajo actual, apunta Ferrer, «es el primer paso para resolver estos problemas». «Han creado un sistema complejo que permite cotejar la secuencia genómica de una persona con todas estas posibles secuencias humanas, en lugar de con una sola secuencia y el consorcio tiene planes de desarrollar esta estrategia con la secuencia de muchos más individuos».
Para José Manuel Castro Tubío, líder del Grupo de Investigación de Genomas y Enfermedad del Centro de Investigación en Medicina Molecular y Enfermedades Crónicas (CIMUS) de Santiago, este nuevo recurso va a ayudar, en primer lugar, a «conocer mejor nuestra identidad, a conocer qué es lo que nos hace diferentes genéticamente a unos de otros». Y el hecho de «conocer lo que nos hace diferentes, qué secuencias de material genético nos hacen diferentes, nos va a permitir saber cosas acerca de nuestra evolución y nos va a permitir también saber cosas sobre las enfermedades genéticas que nos afectan».
«La variabilidad genética va asociada a rasgos biológicos y también a la predisposición de desarrollar enfermedades», explica. «Estos nuevos genomas que ahora se publican van a permitir descubrir muchas variantes que todavía no sabemos a qué se asocian».
Si bien la nueva referencia presentada todavía es un primer borrador y solo representa a un número todavía pequeño de individuos, contiene información que va a ser muy útil para avanzar en la investigación biomédica, concluye el investigador.
Mayo 10/2023 (Diario Médico) – Tomado de Genética – Investigación Copyright Junio 2018 Unidad Editorial Revistas, S.L.U. Todos los derechos reservados
abr
27
En un descubrimiento que podría mejorar la supervivencia de los pacientes con cáncer de vejiga, científicos de Northwestern Medicine han desarrollado una prueba de firma de biomarcadores para predecir qué tumores responderán a la inmunoterapia.
Los fármacos de inmunoterapia de punto de control, que activan el sistema inmunitario del organismo para que reconozca un tumor, sólo son eficaces en un 20% de los casos de cáncer de vejiga. Pero los clínicos desconocen qué pacientes se beneficiarán y por qué no son más eficaces para todos.
En el nuevo estudio -con múltiples colaboradores internacionales- los investigadores de la Facultad de Medicina Feinberg de la Universidad Northwestern identificaron tres tipos de tumores que podrían responder a la inmunoterapia y dos que no. Mediante una combinación de perfiles de expresión génica, mutaciones y proteómica espacial, los científicos también analizaron los cánceres que no respondían para identificar posibles nuevos fármacos y terapias que pudieran utilizarse para hacerlos sensibles a la inmunoterapia.
El estudio se publicará el 27 de abril en Nature Communications.
El cáncer de vejiga, suele ser letal y no ha experimentado mejoras en la supervivencia en los últimos 30 años.
«La inmunoterapia ha cambiado la forma de tratar el cáncer de vejiga, pero tiene importantes limitaciones, ya que la mayoría de los pacientes no responden al tratamiento», afirma el Dr. Joshua Meeks, investigador principal, profesor asociado de urología en Feinberg y urólogo de Northwestern Medicine. «A miles de pacientes se les extirpa la vejiga cada año, y tratar a estos pacientes con inmunoterapia podría mejorar la supervivencia y aumentar potencialmente sus posibilidades de conservar la vejiga en lugar de extirpársela quirúrgicamente».
En este estudio, los investigadores comenzaron con un ensayo de fase II de 82 pacientes tratados con Keytruda (una inmunoterapia) antes de la extirpación de la vejiga. Se trataba de un ensayo único que evaluaba el perfil de expresión génica antes y después de Keytruda, con la capacidad de medir completamente la respuesta a Keytruda cuando se extirpaba la vejiga. Normalmente, Keytruda y otras inmunoterapias se utilizan en pacientes con cáncer metastásico, y los cambios biológicos que se producen en el tumor no se pueden monitorizar con biopsias tumorales. Mediante el perfil del transcriptoma, las alteraciones del ADN y los cambios espaciales que se producían en los tumores tratados con Keytruda, los investigadores pudieron identificar qué características se asociaban con la respuesta o la resistencia.
Por ejemplo, un tercio de los tumores pertenecían a un subtipo con muy pocas células inmunitarias infiltradas en el tumor, pero con una mayor expresión de la vía oncogénica regulada por FGFR3 y una nueva red reguladora de genes activados por un regulador epigenético KDM5B. Dirigiéndose al FGFR3 o al KDM5B, los investigadores podrían volver a despertar una respuesta inmunitaria. Dentro de un año se iniciarán en Northwestern Medicine ensayos clínicos con nuevas combinaciones de medicamentos e inmunoterapia para superar la resistencia inmunitaria.
Además, las conclusiones del estudio también proporcionan «un atlas genómico del cáncer más funcional», afirmó Meeks, que también es catedrático de Urología Edward Schaeffer, M.D., Ph.D. y miembro del Centro Oncológico Integral Robert H. Lurie de la Universidad Northwestern. «El atlas genómico actual examina los componentes fundamentales del cáncer de vejiga, pero no describe ningún tratamiento. Eso es lo que hace significativa esta evaluación. Se trata de cómo responde el cáncer de vejiga a la inmunoterapia».
El Atlas del Genoma del Cáncer es un programa de genómica del cáncer del Instituto Nacional del Cáncer que caracterizó molecularmente más de 20.000 cánceres primarios. El autor principal de este trabajo, el Dr. A. Gordon Robertson, fue también investigador principal del atlas genómico del cáncer de vejiga.
Los hallazgos fueron el resultado de múltiples colaboraciones con grupos internacionales que podían realizar ensayos con inmunoterapia que no estaban disponibles en EE.UU. «Gracias a los esfuerzos científicos en equipo, pudimos aprovechar distintos conocimientos especializados y muestras poco comunes de ensayos clínicos para responder a preguntas importantes sobre qué pacientes responderán a la inmunoterapia para el tratamiento del cáncer de vejiga», afirmó Meeks.
Los científicos de Northwestern colaboraron con investigadores clínicos principales de Italia (Dr. Andrea Necchi) y el Reino Unido (Dr. Thomas Powles.) para desarrollar un biomarcador con bioinformáticos de Canadá y Francia (Clarice Groeneveld). A continuación, los investigadores validaron este biomarcador en una tercera cohorte que trataba a pacientes de todo el mundo.
La investigación se financió principalmente con fondos del Polsky Urologic Cancer Institute del Robert H. Lurie Comprehensive Cancer Center de la Northwestern University en el Northwestern Memorial Hospital, la AACR-Bayer Innovation and Discovery Grant, el Departamento de Defensa de EE.UU. y la Veterans Health Administration.
Abril 27/2023 (MedicalXpress) – Tomado de Immunology Copyright Medical Xpress 2011 – 2023 powered by Science X Network.
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
26
Para el diseño racional de nuevos compuestos materiales, es importante comprender los mecanismos subyacentes a su síntesis. Para estudiar estos mecanismos en las reacciones moleculares suelen emplearse técnicas analíticas como la resonancia magnética nuclear y la espectroscopia. Sin embargo, las vías de reacción que rigen la formación de compuestos cristalinos en estado sólido siguen siendo poco conocidas. Esto se debe en parte a las temperaturas extremas y a las reacciones no homogéneas que se observan en los compuestos en estado sólido. Además, la presencia de numerosos átomos en los compuestos cristalinos sólidos dificulta un análisis preciso. Por tanto, es necesario desarrollar nuevas técnicas que puedan sortear estos retos.
Más recientemente, se han utilizado técnicas de difracción de rayos X (DRX) de sincrotrón in situ para investigar las reacciones que tienen lugar en fases cristalinas. Debido a su alta velocidad y resolución temporal, las medidas de DRX de sincrotrón proporcionan acceso a datos de reacción en ventanas de tiempo extremadamente cortas (unos pocos cientos de milisegundos). Esto hace que la técnica sea prometedora para capturar datos relativos a fases de reacción intermedias de vida corta.
Ahora, un grupo de investigadores japoneses ha utilizado una técnica de DRX de sincrotrón de última generación para estudiar los mecanismos topoquímicos de reducción sólido-gas en la perovskita estratificada. El estudio ha sido dirigido por el profesor asociado Takafumi Yamamoto, del Instituto Tecnológico de Tokio (Tokyo Tech), y publicado en la revista Advanced Science.
«Utilizamos Sr3Fe2O7-δ, una perovskita estratificada de tipo Ruddlesden-Popper, debido a su eficaz capacidad de almacenamiento de oxígeno. El Sr3Fe2O7-δ experimenta reacciones redox topoquímicas reversibles y rápidas bajo O2 y H2 y muestra un excelente rendimiento como material catalizador medioambiental», explica el Dr. Yamamoto.
Sus colaboradores habían observado anteriormente que el dopaje de Sr3Fe2O7-δ con paladio (Pd) aumenta significativamente la velocidad de liberación de oxígeno al tiempo que disminuye la temperatura de liberación. Basándose en estas observaciones, el equipo investigó las vías de reacción y la evolución estructural de esta perovskita durante la reducción sólido-gas.
El equipo comenzó preparando una muestra prístina y una muestra de Sr3Fe2O7-δ cargada con Pd. A continuación, utilizaron DRX de sincrotrón de alta velocidad para monitorizarlas mientras se sometían a una rápida desintercalación de oxígeno (reducción).
Los análisis revelaron que la reducción del Sr3Fe2O7-δ prístino se produjo a través de fases termodinámicamente estables, y que el Sr3Fe2O7-δ prístino experimentó una evolución estructural monofásica gradual durante su reducción. En cambio, la reducción del Sr3Fe2O7-δ cargado de Pd implicó fases intermedias sin equilibrio, una vía drásticamente diferente. Primero se transformó en una fase dinámicamente desordenada durante unos segundos y luego se reorganizó mediante una transición de primer orden para alcanzar el estado final ordenado y estable.
Además, las partículas metálicas de Pd en la superficie del Sr3Fe2O7-δ aceleraron significativamente la reacción de desintercalación de oxígeno del Sr3Fe2O7-δ cargado con Pd en relación con la del Sr3Fe2O7-δ prístino. El Dr. Yamamoto añade: «El cambio en la dinámica de reacción tras la carga de Sr3Fe2O7-δ con Pd demuestra que el tratamiento superficial puede utilizarse para manipular los procesos de reacción en un material cristalino.»
En resumen, estos resultados sugieren que la técnica de DRX de sincrotrón puede aprovecharse para estudiar las vías de reacción en compuestos en estado sólido, así como para identificar sus pasos determinantes de la velocidad. Esto, a su vez, podría ayudar a optimizar la vía de reacción para el diseño racional de materiales funcionales de alto rendimiento.
Abril 26/2023 (EurekaAlert!) – Tomado de News Releases Copyright 2023 by the American Association for the Advancement of Science (AAAS)
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
26
La secuenciación metagenómica (mNGS) es una poderosa herramienta de diagnóstico para detectar patógenos causantes en pruebas microbiológicas clínicas. La clasificación rápida y precisa de las secuencias metagenómicas es un procedimiento crítico para la identificación de patógenos en el paso de laboratorio seco de las pruebas mNGS. Sin embargo, este paso crucial puede mejorarse clasificando las secuencias en un plazo clínicamente relevante.
Para hacer frente a este reto, un equipo de BGI Genomics dirigido por Xuebin Wang ha lanzado recientemente GPMeta, un método ultrarrápido de detección de patógenos, y ha publicado estos aspectos destacados en Briefings in Bioinformatics.
GPMeta puede identificar patógenos de forma rápida y precisa a través de datos de secuenciación mNGS complejos y masivos. Utilizando conjuntos de datos simulados y conjuntos de datos de secuenciación metagenómica de muestras clínicas, los resultados se compararon con herramientas utilizadas por la comunidad de investigadores bioinformáticos como Bowtie2, Bwa, Kraken2 y Centrifuge.
Los resultados muestran que GPMeta no sólo tiene una mayor precisión, sino que también exhibe una velocidad con un incremento de velocidad significativo. Además, GPMeta ofrece un algoritmo de agrupamiento GPMetaC, un modelo estadístico para agrupar y volver a puntuar alineaciones ambiguas con el fin de mejorar la discriminación de secuencias altamente homólogas de genomas microbianos con una identidad nucleotídica media >95%. Estos resultados subrayan el papel clave de GPMeta en el desarrollo de la prueba mNGS en enfermedades infecciosas que requieren tiempos de respuesta rápidos.
Antecedentes
La detección más rápida y temprana de los patógenos causantes es fundamental para una terapia antibiótica precisa en lugar de un tratamiento empírico. Puede detectar simultáneamente casi todos los microorganismos patógenos nuevos y conocidos en el cuerpo del paciente en una sola prueba y tiene enormes aplicaciones potenciales en el diagnóstico de infecciones.
La detección de mNGS consta de dos componentes: las manipulaciones experimentales en laboratorio húmedo, que incluyen el preprocesamiento de muestras clínicas, la extracción de ácido nucleico total, la preparación de bibliotecas y la secuenciación, y el análisis bioinformático en laboratorio seco, que incluye el preprocesamiento de datos de secuenciación brutos, la eliminación de secuencias de huéspedes humanos, la alineación de secuencias con la base de datos curada de patógenos y la clasificación taxonómica de secuencias microbianas.
El análisis bioinformático es el último paso crucial en la detección de mNGS, que debe completarse con rapidez y precisión para acelerar todo el proceso de detección. Sin embargo, existe una necesidad urgente de nuevas estrategias para acelerar el análisis bioinformático de la identificación de patógenos.
Para hacer frente a este reto, GPMeta utiliza un esquema de índice hash sucinto y admite múltiples GPU para llevar a cabo en bases de datos divididas de forma simultánea, lo que satisface una necesidad creciente de la capacidad para hacer frente a un número cada vez mayor de genomas microbianos.
En el conjunto de datos de 25 millones de lecturas, GPMeta y GPMetaC sólo necesitan menos de 3 minutos para completar todo el análisis de detección.
En el conjunto de datos de 110 millones de lecturas (volumen de datos de detección mNGS convencional), GPMeta y GPMetaC sólo necesitan 4 minutos para completar todo el análisis de detección.
Cuando se aplicó a la biblioteca completa de patógenos de 190Gb, GPMeta y GPMetaC la aceleraron 39-50 y 12-35 veces respectivamente en comparación con Bwa y Bowtie2.
La detección completa y el análisis GPMeta son 18 veces y 12 veces más rápidos que Bwa y Bowtie2 respectivamente.
GPMeta admite múltiples GPUs para realizar el alineamiento y la clasificación taxonómica de secuencias microbianas en bases de datos divididas simultáneamente y fusiona automáticamente los resultados de múltiples sub-bases de datos, lo que resulta significativo para mantenerse al día con la rápida expansión de la base de datos de genomas microbianos. Para sacar el máximo partido de GPMeta, es necesario estudiar más a fondo cómo integrarla mejor y con mayor facilidad en las prácticas clínicas.
Abril 26/2023 (EurekaAlert!) – Tomado de News Releases Copyright 2023 by the American Association for the Advancement of Science (AAAS)
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
abr
25
Incluso mientras dormimos, el cerebro no descansa por completo. Sorprendentemente, el flujo sanguíneo en un cerebro dormido puede ser mayor que cuando está en estado de vigilia. Esto permite al cerebro eliminar los metabolitos de desecho, lo que es importante para prevenir el desarrollo y la progresión de disfunciones neurológicas como la demencia. Sin embargo, se desconoce el mecanismo exacto por el que el cerebro dormido aumenta el flujo sanguíneo.
Investigadores dirigidos por el director Kim Seong-Gi, del Centro de Investigación en Neurociencia por Imagen del Instituto de Ciencias Básicas de Corea del Sur, descubrieron recientemente los secretos de este fenómeno. Se descubrió que un tipo de neurona inhibidora llamada «neurona parvalbúmina (PV)» segrega un material llamado «sustancia P» que es responsable de la vasodilatación y el control del flujo sanguíneo al cerebro. El estudio se publica en la revista Proceedings of the National Academy of Sciences.
A diferencia de otras neuronas de subtipo inhibitorio, antes se pensaba que las neuronas PV GABAérgicas no liberaban sustancias vasoactivas, de ahí que su papel en la regulación del flujo sanguíneo haya sido controvertido. Para investigar el papel de las neuronas FV en la regulación del flujo sanguíneo, los investigadores expresaron una proteína opsina llamada canalrodopsina-2 (ChR2) en neuronas FV de ratón, y activaron selectivamente las neuronas FV mediante estimulación luminosa.
Las respuestas vasculares a la activación de las neuronas PV se midieron mediante imágenes ópticas de campo amplio e imágenes de resonancia magnética funcional (fMRI). Además, para identificar el papel de las neuronas PV en el flujo sanguíneo bajo anestesia, los investigadores utilizaron ketamina/xilacina para dormir a los animales.
Los resultados mostraron que, en animales ligeramente anestesiados, la estimulación de las neuronas PV inducía vasoconstricción y una disminución del flujo sanguíneo. A continuación, se producía una vasodilatación lenta y un aumento del flujo sanguíneo desde 20 segundos hasta un minuto después del cese de la estimulación. Por otra parte, en los animales activos, la actividad de las neuronas PV sólo provocó una reducción del flujo sanguíneo. Esto significa que las neuronas FV disponen de dos mecanismos diferentes para controlar el flujo sanguíneo cerebral, dependiendo de si el cerebro está despierto o dormido.
Además, los investigadores también descubrieron el mecanismo que subyace a la vasodilatación lenta observada tras la estimulación optogenética. Cuando se activan las neuronas PV, se inhiben las neuronas excitadoras cercanas, lo que provoca vasoconstricción y reducción del flujo sanguíneo. Al mismo tiempo, se descubrió que estas neuronas PV liberan un péptido denominado «sustancia P», responsable de la vasodilatación lenta observada. La sustancia P activa unas células denominadas neuronas GABAérgicas sintasa de óxido nítrico (nNOS) que segregan óxido nitroso, un conocido vasodilatador.
La presente investigación desvela por fin los factores que controlan el flujo sanguíneo al cerebro durante el sueño, y el papel hasta ahora desconocido de las neuronas PV en este proceso. El director Kim afirmó: «Nuestra investigación sugiere una nueva dirección de investigación sobre los mecanismos de control del flujo sanguíneo cerebral, con posibles implicaciones en el tratamiento del insomnio y los trastornos del sueño.»
Abril 25/2023 (MedicalXpress) – Tomado de Neuroscience Copyright Medical Xpress 2011 – 2023 powered by Science X Network
Traducción realizada con la versión gratuita del traductor www.DeepL.com/Translator
Noticias anteriores a 2010
Suscripción AL Día
