
mar
19
No se encontró ninguna evidencia de que el virus se haya producido en un laboratorio o haya sido diseñado.
Instituto de investigación scripps
El análisis de Scripps Research de los datos públicos de la secuencia del genoma del SARS-CoV-2 y los virus relacionados no encontró evidencia de que el virus se haya producido en un laboratorio o haya sido diseñado.
 El nuevo coronavirus SARS-CoV-2 que surgió en la ciudad de Wuhan, China, el año pasado y que desde entonces ha causado una epidemia de COVID-19 a gran escala y se ha extendido a más de 70 países, es producto de la evolución natural, según los resultados publicados en la revista Nature Medicine.
El nuevo coronavirus SARS-CoV-2 que surgió en la ciudad de Wuhan, China, el año pasado y que desde entonces ha causado una epidemia de COVID-19 a gran escala y se ha extendido a más de 70 países, es producto de la evolución natural, según los resultados publicados en la revista Nature Medicine.
El análisis de los datos públicos de la secuencia del genoma del SARS-CoV-2 y los virus relacionados no encontró evidencia de que el virus se haya producido en un laboratorio o haya sido diseñado de otro modo.
«Al comparar los datos disponibles de la secuencia del genoma para las cepas conocidas de coronavirus, podemos determinar firmemente que el SARS-CoV-2 se originó a través de procesos naturales«, dijo Kristian Andersen, PhD, profesor asociado de inmunología y microbiología en Scripps Research y autor correspondiente en el papel.
Además de Andersen, los autores del artículo, «El origen próximo del SARS-CoV-2«, incluyen a Robert F. Garry, de la Universidad de Tulane; Edward Holmes, de la Universidad de Sydney; Andrew Rambaut, de la Universidad de Edimburgo; W. Ian Lipkin, de la Universidad de Columbia.
Los coronavirus son una gran familia de virus que pueden causar enfermedades que varían ampliamente en severidad. La primera enfermedad grave conocida causada por un coronavirus surgió con la epidemia del Síndrome Respiratorio Agudo Severo (SRAS) de 2003 en China. Un segundo brote de enfermedad grave comenzó en 2012 en Arabia Saudita con el Síndrome Respiratorio del Medio Oriente (MERS).
El 31 de diciembre del año pasado, las autoridades chinas alertaron a la Organización Mundial de la Salud sobre un brote de una nueva cepa de coronavirus que causa una enfermedad grave, que posteriormente se denominó SARS-CoV-2. Hasta el 20 de febrero de 2020, se han documentado casi 167 500 casos de COVID-19, aunque es probable que muchos casos más leves no hayan sido diagnosticados. El virus ha matado a más de 6 600 personas.
Poco después de que comenzara la epidemia, los científicos chinos secuenciaron el genoma del SARS-CoV-2 y pusieron los datos a disposición de los investigadores de todo el mundo.
Los datos de la secuencia genómica resultante han demostrado que las autoridades chinas detectaron rápidamente la epidemia y que el número de casos de COVID-19 ha aumentado debido a la transmisión de humano a humano después de una sola introducción en la población humana. Andersen y sus colaboradores en varias otras instituciones de investigación utilizaron estos datos de secuenciación para explorar los orígenes y la evolución del SARS-CoV-2 al enfocarse en varias características reveladoras del virus.
Los científicos analizaron la plantilla genética para las proteínas espiga, armaduras en el exterior del virus que utiliza para atrapar y penetrar las paredes externas de las células humanas y animales. Más específicamente, se centraron en dos características importantes de la proteína espiga: el dominio de unión al receptor (RBD), un tipo de gancho de agarre que se adhiere a las células huésped, y el sitio de escisión, un abridor de latas molecular que permite que el virus se abra e ingresar a las células anfitrionas.
Evidencia de evolución natural
Los científicos descubrieron que la porción RBD de las proteínas de la punta del SARS-CoV-2 había evolucionado para atacar efectivamente una característica molecular en el exterior de las células humanas llamada ACE2, un receptor involucrado en la regulación de la presión arterial. La proteína del pico SARS-CoV-2 fue tan efectiva en la unión de las células humanas, de hecho, que los científicos concluyeron que era el resultado de la selección natural y no el producto de la ingeniería genética.
Esta evidencia de evolución natural fue respaldada por datos sobre la columna vertebral del SARS-CoV-2: su estructura molecular general. Si alguien buscara diseñar un nuevo coronavirus como patógeno, lo habría construido a partir de la columna vertebral de un virus que se sabe que causa enfermedades. Pero los científicos descubrieron que la columna vertebral del SARS-CoV-2 difería sustancialmente de las de los coronavirus ya conocidos y en su mayoría se parecían a virus relacionados que se encuentran en murciélagos y pangolines.
«Estas dos características del virus, las mutaciones en la porción RBD de la proteína espiga y su columna vertebral distinta, descartan la manipulación de laboratorio como un posible origen del SARS-CoV-2«, dijo Andersen.
Josie Golding, PhD, líder de epidemias en Wellcome Trust, con sede en el Reino Unido, dijo que los hallazgos de Andersen y sus colegas son «crucialmente importantes para aportar una visión basada en la evidencia de los rumores que han estado circulando sobre los orígenes del virus (SARS-CoV) -2) causando COVID-19 «.
«Concluyen que el virus es producto de la evolución natural», agrega Goulding, «poniendo fin a cualquier especulación sobre ingeniería genética deliberada». (dar doble clik en la imagen para verla ampliada
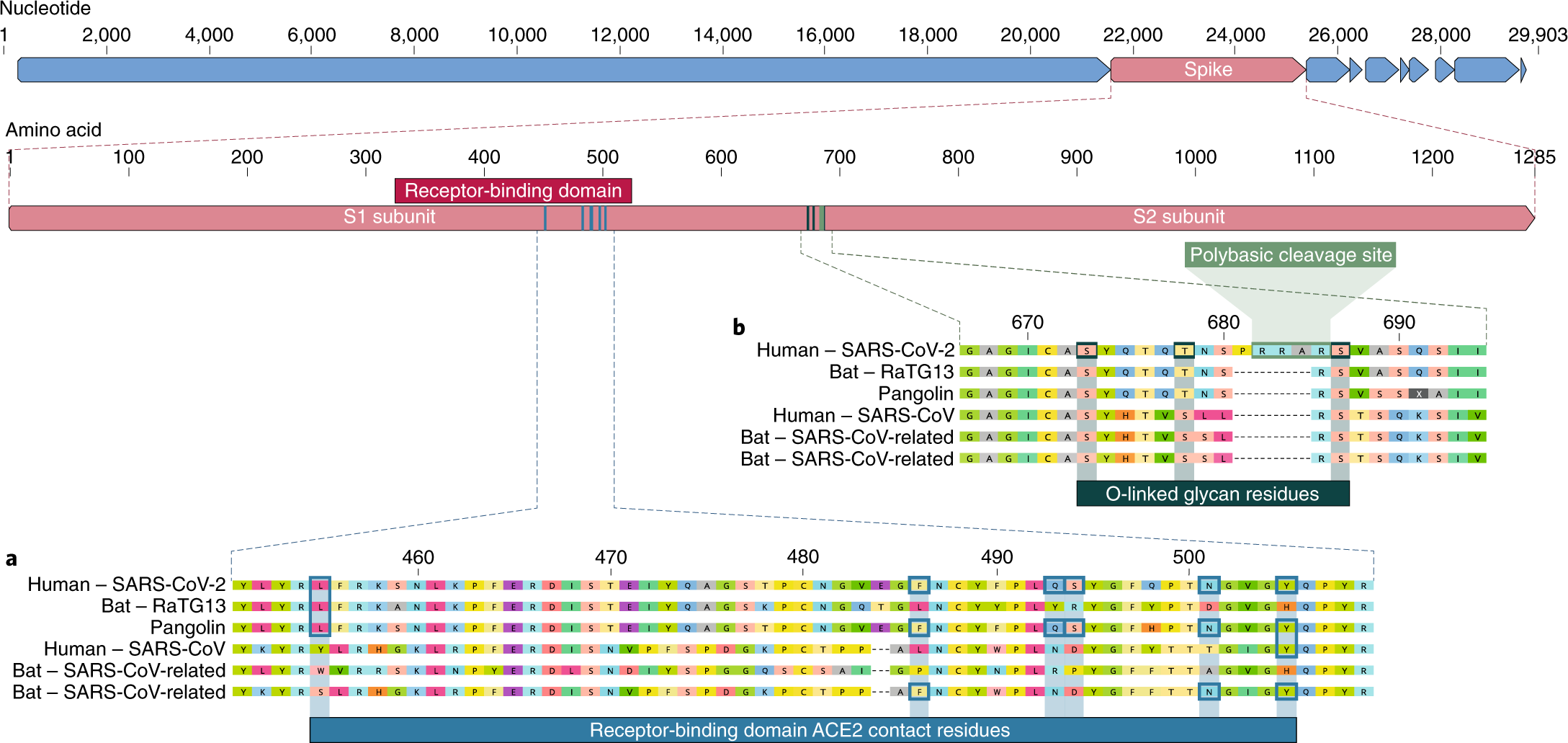 a, Mutaciones en los residuos de contacto de la proteína espiga de SARS-CoV-2. La proteína espiga del SARS-CoV-2 (barra roja en la parte superior) se alineó contra los coronavirus similares al SARS-CoV más estrechamente relacionados y el propio SARS-CoV. Los residuos clave en la proteína espiga que hacen contacto con el receptor ACE2 están marcados con recuadros azules tanto en el SARS-CoV-2 como en los virus relacionados, incluido el SARS-CoV (cepa Urbani). b, Adquisición del sitio de escisión polibásica y glicanos unidos a O. Tanto el sitio de escisión polibásico como los tres glicanos unidos a O pronosticados adyacentes son exclusivos del SARS-CoV-2 y no se vieron previamente en los betacoronavirus de linaje B. Las secuencias que se muestran son de NCBI GenBank, códigos de acceso MN908947, MN996532, AY278741, KY417146 y MK211376. Las secuencias de coronavirus de pangolín son un consenso generado a partir de SRR10168377 y SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
a, Mutaciones en los residuos de contacto de la proteína espiga de SARS-CoV-2. La proteína espiga del SARS-CoV-2 (barra roja en la parte superior) se alineó contra los coronavirus similares al SARS-CoV más estrechamente relacionados y el propio SARS-CoV. Los residuos clave en la proteína espiga que hacen contacto con el receptor ACE2 están marcados con recuadros azules tanto en el SARS-CoV-2 como en los virus relacionados, incluido el SARS-CoV (cepa Urbani). b, Adquisición del sitio de escisión polibásica y glicanos unidos a O. Tanto el sitio de escisión polibásico como los tres glicanos unidos a O pronosticados adyacentes son exclusivos del SARS-CoV-2 y no se vieron previamente en los betacoronavirus de linaje B. Las secuencias que se muestran son de NCBI GenBank, códigos de acceso MN908947, MN996532, AY278741, KY417146 y MK211376. Las secuencias de coronavirus de pangolín son un consenso generado a partir de SRR10168377 y SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
Posibles orígenes del virus
Con base en su análisis de secuenciación genómica, Andersen y sus colaboradores concluyeron que los orígenes más probables para el SARS-CoV-2 siguieron uno de los dos escenarios posibles.
En un escenario, el virus evolucionó a su estado patógeno actual a través de la selección natural en un huésped no humano y luego saltó a los humanos. Así es como han surgido brotes previos de coronavirus, con humanos contrayendo el virus después de la exposición directa a civetas (SARS) y camellos (MERS). Los investigadores propusieron a los murciélagos como el reservorio más probable para el SARS-CoV-2, ya que es muy similar a un coronavirus de murciélago. Sin embargo, no hay casos documentados de transmisión directa murciélago-humano, lo que sugiere que un huésped intermedio probablemente estuvo involucrado entre murciélagos y humanos.
En este escenario, las dos características distintivas de la proteína espiga del SARS-CoV-2, la porción RBD que se une a las células y el sitio de escisión que abre el virus, habrían evolucionado a su estado actual antes de ingresar a los humanos. En este caso, la epidemia actual probablemente habría surgido rápidamente tan pronto como los humanos estuvieran infectados, ya que el virus ya habría desarrollado las características que lo hacen patógeno y capaz de propagarse entre las personas.
En el otro escenario propuesto, una versión no patógena del virus saltó de un huésped animal a humanos y luego evolucionó a su estado patógeno actual dentro de la población humana. Por ejemplo, algunos coronavirus de pangolines, mamíferos tipo armadillo que se encuentran en Asia y África, tienen una estructura RBD muy similar a la del SARS-CoV-2. Un coronavirus de un pangolín podría haberse transmitido a un humano, ya sea directamente o a través de un huésped intermediario, como civetas o hurones.
Entonces, la otra proteína de espiga característica del SARS-CoV-2, el sitio de escisión, podría haber evolucionado dentro de un huésped humano, posiblemente a través de una circulación limitada no detectada en la población humana antes del comienzo de la epidemia.
Los investigadores encontraron que el sitio de escisión del SARS-CoV-2 parece similar a los sitios de escisión de cepas de gripe aviar que se ha demostrado que se transmite fácilmente entre las personas. El SARS-CoV-2 podría haber desarrollado un sitio de escisión tan virulento en las células humanas y pronto inició la epidemia actual, ya que el coronavirus posiblemente se habría vuelto mucho más capaz de propagarse entre las personas.
El coautor del estudio, Andrew Rambaut, advirtió que es difícil, si no imposible, saber en este momento cuál de los escenarios es más probable. Si el SARS-CoV-2 ingresó a los humanos en su forma patógena actual de una fuente animal, aumenta la probabilidad de brotes futuros, ya que la cepa del virus que causa la enfermedad aún podría estar circulando en la población animal y podría volver a saltar humanos. Las posibilidades son menores que, un coronavirus no patógeno entre en la población humana y luego desarrolle propiedades similares al SARS-CoV-2.
Referencia Bibliográfica:
Kristian G. Andersen, Andrew Rambaut, W. Ian Lipkin, Edward C. Holmes & Robert F. Garry . The proximal origin of SARS-CoV-2. Nature Medicine. DOI https://doi.org/10.1038/s41591-020-0820-9
Comments
Noticias anteriores a enero de 2010
Suscripción AL Día
