
abr
21
 Hasta un 10 % de las personas que padecen sequedad ocular desconoce esta condición, que sucede cuando los ojos no se encuentran bien lubricados y se generan molestias como picor, escozor, ojo rojo, lagrimeo y molestias visuales, afectando además a uno de cada tres españoles.
Hasta un 10 % de las personas que padecen sequedad ocular desconoce esta condición, que sucede cuando los ojos no se encuentran bien lubricados y se generan molestias como picor, escozor, ojo rojo, lagrimeo y molestias visuales, afectando además a uno de cada tres españoles.
De cara a minimizar esta cifra, la asociación Visión y Vida y el Fórum Contactología han creado una breve encuesta virtual que utilizarán los profesionales ópticos-optometristas en gabinete durante el mes de abril, pero que también podrá realizar cualquier usuario a través de Internet para ver si padece o no sequedad ocular.
En la encuesta se realizan preguntas sobre cierta sintomatología de sequedad ocular como la sensibilidad a la luz, la existencia de sensación de arenilla o dolor en los ojos, visión borrosa o mala visión, así como la dificultad para realizar ciertas acciones básicas como son la lectura, la conducción nocturna, el uso de pantallas o ver la televisión.
El presidente de Visión y Vida, Salvador Alsina, ha explicado que los resultados pueden mostrar si se trata de un ojo seco leve, moderado o severo y que, las personas que lo hayan hecho por su cuenta, podrán basarse en ellos para acercarse a un óptico-optometrista de confianza para que le asesore y le explique cómo paliar su problema visual y disfrutar de una visión confortable.
«OSDI es un sencillo cuestionario que permite clasificar la gravedad de la sequedad ocular que se padece según la sintomatología presentada y la intensidad de la misma», ha añadido Alsina de cara al Día Mundial de las Lentillas, que se celebra este martes.
En el marco de este día, ambas organizaciones buscan informar sobre las ventajas del uso de lentes de contacto y mitigar problemas que acaban generando rechazo a la hora de usarlas, para lo que realizarán actividades en establecimientos sanitarios de óptica y a través de redes sociales para explicar los usos y beneficios que tiene el incorporar este, apoyándose en la figura de Leonardo da Vinci.
«Hablamos de esa pequeña gran idea de Leonardo da Vinci que ha cambiado el mundo de la salud visual», ha resaltado Alsina, recordando que el inventor está considerado como el precursor de las lentes de contacto por introducir la cabeza en un recipiente con agua y observar cómo cambiaba la refracción de la luz y modificaba la visión, que sirvió como base para dar con la creación de las lentillas.
14 abril 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
18
 «Estamos viendo un incremento importante, casi hasta el doble, del número de menores de edad con miopía«, ha explicado el doctor Fernández Perianes, quien ha achacado esta situación al exceso de iluminación de los dispositivos y a su cercanía a los ojos.
«Estamos viendo un incremento importante, casi hasta el doble, del número de menores de edad con miopía«, ha explicado el doctor Fernández Perianes, quien ha achacado esta situación al exceso de iluminación de los dispositivos y a su cercanía a los ojos.
El especialista ha resaltado que los niños que usan dispositivos móviles comienzan a manifestar una pérdida de visión lejana a partir de los seis o siete años, lo que se debe a un uso prolongado de la visión cercana, lo que evita que el ojo se desarrolle en condiciones «óptimas» para tener una buena visión de lejos.
Un menor desarrollo del aparato visual se acaba manifestando en síntomas como enrojecimiento, ojo seco, visión borrosa, dolor de cabeza y molestias en los párpados.
«Hay que tener en cuenta que los móviles lo que están haciendo es emitir una luz intensa en un ojo que todavía no está preparado para ello, por lo que esa cantidad de iluminación que está recibiendo en una distancia muy corta supone que el riesgo de miopía sea mucho más elevado», ha insistido.
Fernández Perianes ha señalado que la «única medida efectiva» para no llegar a esta situación es evitar que los menores usen dispositivos móviles o restringir su uso «considerablemente», unas medidas avaladas por la Organización Mundial de la Salud (OMS) y diferentes sociedades oftalmológicas y pediátricas.
Aunque las recomendaciones difieren dependiendo de la edad y de las horas de uso, los expertos están de acuerdo en que el uso de pantallas durante los tres primeros años de vida debe ser inexistente, un límite que puede llegar a marcarse en edades más altas, pues a menor tiempo de exposición a las pantallas, menor será la probabilidad de desarrollar problemas oculares.
«Hasta los 16 años, es importante hacer un uso responsable y fijar límites de tiempo, siempre con dispositivos analógicos o sin conexión a Internet, ya que esto, entre otros muchos problemas, fideliza mucho más al menor a la pantalla, haciendo un daño mayor a sus ojos», ha añadido.
En caso de que aparezca algún síntoma, el doctor ha recomendado acudir a un oftalmólogo para revisar si se tiene algún defecto de refracción, además de aconsejar una lubricación del ojo con lágrimas artificiales, usar una buena iluminación y evitar los reflejos.
Del mismo modo, ha subrayado que la única corrección de esta patología es a través del uso de las gafas, pues hasta que el ojo no concluya su desarrollo, que suele ser a los 20 o 21 años, no se pueden valorar opciones quirúrgicas.
«A ver si llega el momento en que todos nos concienciamos de que los niños pequeños no deben utilizar dispositivos si no se quieren ver abocados luego a problemas refractivos, que en muchos casos ya son irremediables como no sean como cirugía», ha concluido.
14 abril 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
dic
30
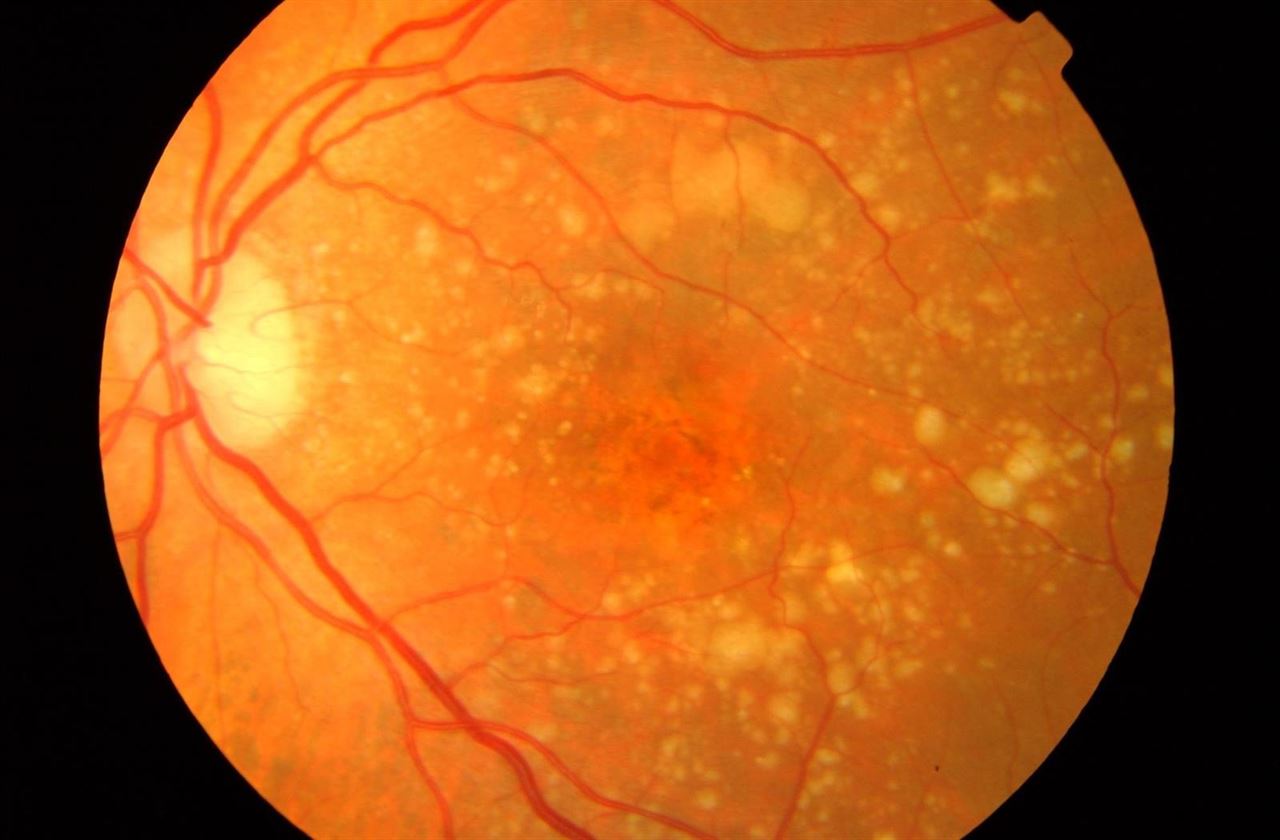 La degeneración macular asociada a la edad (DMAE) es la principal causa de pérdida de visión en personas mayores de 65 años, y se caracteriza por cambios anormales en la mácula que provocan una reducción de la visión y distorsión de los objetos. La DMAE seca representa el 90 % de todos los casos de DMAE, con un deterioro de la visión relativamente leve; sin embargo, aproximadamente el 30 % evoluciona hacia la pérdida grave de visión asociada a la DMAE húmeda en un plazo de 10 años.
La degeneración macular asociada a la edad (DMAE) es la principal causa de pérdida de visión en personas mayores de 65 años, y se caracteriza por cambios anormales en la mácula que provocan una reducción de la visión y distorsión de los objetos. La DMAE seca representa el 90 % de todos los casos de DMAE, con un deterioro de la visión relativamente leve; sin embargo, aproximadamente el 30 % evoluciona hacia la pérdida grave de visión asociada a la DMAE húmeda en un plazo de 10 años.
Los únicos tratamientos aprobados por la Administración de Alimentos y Medicamentos de Estados Unidos (FDA, por sus siglas en inglés) para la DMAE seca a partir de 2023 son dos fármacos inyectables, limitados por la preocupación que suscitan las complicaciones de las inyecciones intravítreas y su modesta eficacia para restaurar la visión.
Los colirios son el método de administración de fármacos preferido en el mercado oftálmico, pero el desarrollo de fórmulas dirigidas a la retina, situada en el segmento posterior del ojo, sigue siendo un reto importante.
Para abordar las limitaciones de los tratamientos basados en inyecciones, el equipo de investigación, que ha publicado su trabajo en la revista Advanced Science, se centró en la vía de señalización inflamatoria de los receptores tipo Toll (TLR), de los que se sabe que desempeñan un papel fundamental en la patogénesis de la DMAE.
Extrayendo secuencias peptídicas de decenas de miles de proteínas con estructuras similares a las proteínas de señalización TLR naturales, crearon una extensa biblioteca de más de 190 000 péptidos candidatos a fármacos. Utilizando tecnología avanzada para el cribado rápido de péptidos que se unen específicamente a las proteínas de señalización TLR, identificaron con éxito múltiples péptidos candidatos capaces de inhibir las interacciones entre estas proteínas.
Los investigadores validaron la eficacia terapéutica de los péptidos administrándolos como colirios a ratones con DMAE seca inducida. El grupo tratado mostró una protección de las células de la retina y una reducción significativa de la degeneración retiniana, comparable a la de los ratones normales. Esto demostró que los colirios basados en péptidos podrían sustituir eficazmente a las terapias inyectables existentes para la DMAE seca.
MAYOR COMODIDAD
Este nuevo agente terapéutico, administrado en forma de gotas oftálmicas, ofrece a los pacientes una mayor comodidad y adherencia al tratamiento, al tiempo que reduce las complicaciones y los costes asociados a los tratamientos invasivos repetitivos. Además, la naturaleza no invasiva y segura de la terapia proporciona una nueva opción de tratamiento que mejora tanto la eficacia como la satisfacción del paciente. Se espera que esta innovación revolucione la accesibilidad al tratamiento de la DMAE y otras afecciones oftálmicas relacionadas.
«El Centro de Desarrollo de Fármacos de Productos Naturales de Corea (KIST), creado en septiembre para centrarse en la investigación orientada a la misión, tiene como objetivo desarrollar fármacos globales dirigidos a enfermedades relacionadas con el envejecimiento, incluidos el cáncer y las afecciones oftalmológicas. Tenemos previsto seguir investigando en colaboración con empresas farmacéuticas nacionales e internacionales para avanzar en los ensayos clínicos mundiales de esta innovadora terapia para la DMAE seca», ha finalizado el doctor Moon-Hyeong Seo que dirige el equipo de investigación del KIST.
26 diciembre 2024|Fuente: Europa Press |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2024. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
dic
25
 Cuba revisa hoy diversas iniciativas en pos de elevar el nivel de los servicios sanitarios en Haití, donde la mayor de las Antillas mantiene una brigada de colaboradores de la salud desde hace 26 años.
Cuba revisa hoy diversas iniciativas en pos de elevar el nivel de los servicios sanitarios en Haití, donde la mayor de las Antillas mantiene una brigada de colaboradores de la salud desde hace 26 años.
Durante una reunión efectuada en esta capital, el ministro de Salud Pública y Población de Haití, Duckenson Lorthe, y el embajador de Cuba en este país caribeño, Carlos Moya, destacaron el significativo aporte del contingente cubano, el cual responde a las crisis del sector, ofreciendo su atención en regiones a menudo desatendidas.
Un tema importante discutido fue el aumento en el número de becas otorgadas a estudiantes haitianos, especialmente para las especialidades médicas más difícil de ver en el país.
Esta iniciativa tiene como objetivo formar a una nueva generación de profesionales de la salud, calificados que puedan satisfacer las crecientes necesidades del sistema asistencial haitiano.
El diario Haití Libre destacó que en 26 años la Brigada Médica de Cuba en Haití realizó 36 167 339 consultas, 200 827 partos, 22 582 por cesáreas, 725 093 intervenciones quirúrgicas, de ellas, 249 318 mayores, y 210 100 pacientes fueron rehabilitados en los servicios de fisioterapia.
Con la Operación Milagro, para mejorar la visión de los haitianos, fueron intervenidos por los oftalmólogos cubanos 73 404 personas.
En cuanto a los exámenes de laboratorio fueron realizados un 1 270 416, 39 034 radiografías, 745 651 ultrasonidos, 114 996 endoscopias, 115 309 electrocardiogramas y un total de 77 391 vidas salvadas.
23 diciembre 2024|Fuente: Prensa Latina |Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2024. Agencia Informativa Latinoamericana Prensa Latina S.A.|Noticia
dic
3
 Trabajadores del sector de la Salud de Cuba y la región celebran hoy el Día de la Medicina Latinoamericana, en homenaje al natalicio del científico cubano Carlos Juan Finlay y Barrés (1833-1915), descubridor del agente transmisor de la fiebre amarilla.
Trabajadores del sector de la Salud de Cuba y la región celebran hoy el Día de la Medicina Latinoamericana, en homenaje al natalicio del científico cubano Carlos Juan Finlay y Barrés (1833-1915), descubridor del agente transmisor de la fiebre amarilla.
El doctor Finlay, el más profundo e intenso investigador de esta enfermedad, concluyó que entre un sujeto infectado y otro sano, había un agente independiente que la transmitía, y fue capaz de identificar al Aedes aegypti como el vector biológico, resalta el sitio web de la Organización Panamericana de la Salud.
Su victoria quiso ser escamoteada por los Estados Unidos para favorecer al norteamericano Walter Reed, quien presidió, en 1901, la cuarta comisión estadounidense que vino a Cuba, precisamente, para «demostrar» in situ que la fiebre amarilla tenía un origen bacteriano y que, por tanto, Finlay estaba equivocado.
El médico cubano había estado en aquel país en febrero de 1881 para presentar su trabajo «El mosquito considerado hipotéticamente como agente de la transmisión de la fiebre amarilla», y había sido ignorado.
Sin embargo, la oposición a reconocer a Reed como el verdadero descubridor se puso de manifiesto cuando Francia decide otorgar a Finlay en 1911 la orden oficial de la Legión de Honor, e Inglaterra la medalla Mary Kinsley, concedida en el mundo solo a los científicos Mauson, Ross y al genial Koch, descubridor del bacilo de la tuberculosis.
Asimismo, el XIV Congreso Internacional de Historia de la Medicina, celebrado en Roma en 1954, ratificó al cubano como el único descubridor del agente trasmisor de la fiebre amarilla y la aplicación de su doctrina en el saneamiento del trópico.
Dos años después, esta misma cita, realizada en España, acordó la ejecución de una campaña intensa para que los libros de texto, diccionarios enciclopédicos y medios de divulgación no atribuyeran a otras personas la gloria que, por derecho propio, le pertenecía.
Finlay fue propuesto siete veces para el Premio Nóbel de Medicina, pero los Estados Unidos siempre se opusieron. En la década del cincuenta, por fin se esclarece la verdad histórica y se instaura el Día de la Medicina Latinoamericana en reconocimiento al cubano.
El 25 de mayo de 1981 la Unesco instituyó por primera vez el Premio Internacional Carlos J. Finlay, para reconocer avances en Microbiología, e incluyó al sabio en su revista como uno de los seis microbiólogos más destacados de la historia mundial.
Para conmemorar esta fecha, en Cuba se desarrolla una Jornada de Homenaje al Trabajador de la Salud, que inicia el 22 de noviembre con el Día del farmacéutico.
La víspera, en la sede del Ministerio de Salud Pública (Minsap), fueron reconocidos profesionales que se crecieron con sobradas muestras de consagración y compromiso, durante la contingencia electroenergética que enfrentó el país y en la etapa de recuperación tras el paso de los huracanes Oscar y Rafael, destaca la página web del Minsap.
Este lunes conmemoramos en el @MINSAPCuba el Día de la Medicina Latinoamericana, donde reconocimos a trabajadores destacados de nuestro centro, por su entrega en tiempos tan complejos, con multiplicados desafíos. ¡Gracias a todos por acompañar siempre la defensa de la vida!, escribió en su cuenta en X el ministro José Ángel Portal.
03 diciembre 2024|Fuente: Prensa Latina |Tomado de |Noticia
oct
11
 India eliminó el tracoma, una infección bacteriana que afecta los ojos, como un problema de salud pública, convirtiéndose así en el tercer país de la región en alcanzar este hito, trascendió hoy aquí.
India eliminó el tracoma, una infección bacteriana que afecta los ojos, como un problema de salud pública, convirtiéndose así en el tercer país de la región en alcanzar este hito, trascendió hoy aquí.
Saima Wazed, directora regional de la Organización Mundial de la Salud (OMS), entregó en Nueva Delhi la certificación oficial que acredita ese logro a Aradhana Patnaik, funcionaria del Ministerio de Salud y Bienestar Familiar.
Causada por la bacteria Chlamydia trachomatis, el tracoma es contagiosa y se propaga a través del contacto con los ojos, párpados, nariz o secreciones de la garganta de personas infectadas y si no se trata, causa ceguera irreversible.
La OMS la calificó como una enfermedad tropical desatendida, estimó que 150 millones de personas en todo el mundo están afectadas por la dolencia y seis millones de ellas son ciegas o corren el riesgo de sufrir complicaciones que les provoquen discapacidad visual.
Su incidencia es mayor en comunidades desfavorecidas que viven en malas condiciones ambientales.
La enfermedad fue una de las principales causas de ceguera en India durante en las décadas de 1950 y 1960, lo que obligó al Gobierno lanzar el Programa Nacional de Control del Tracoma en 1963 y posteriormente los esfuerzos por controlarla se integraron en el Programa Nacional de Control de la Ceguera de la India.
En 1971, la ceguera debido al tracoma era del 5 % y hoy, gracias a las diversas iniciativas gubernamentales se redujo a menos del 1 %, indicó el Ministerio de Salud Pública.
De igual modo, la OMS contribuyó con los satisfactorios resultados con la estrategia SAFE que significa adopción de cirugía, antibióticos, higiene facial, limpieza ambiental, entre otras medidas.
Como resultado, en 2017, India fue declarada libre de tracoma infeccioso; sin embargo, la vigilancia de los casos continuó en todos los distritos desde 2019 hasta 2024.
Entre 2021 y 2024, la Encuesta Nacional de Triquiasis Tracomatosa en 200 distritos endémicos corroboró el logro sanitario que posibilitó a la OMS declarar a India libre del tracoma como un problema de salud pública.
09 octubre 2024|Fuente: Prensa Latina |Tomado de |Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
