
sep
18
 Un estudio pionero de la Universidad de Granada (UGR), en el sur de España, y el Hospital Universitario de Düsseldorf (Alemania) ha abierto una nueva vía terapéutica para enfermedades metabólicas pediátricas y ha permitido desarrollar un medicamento huérfano -destinado al diagnóstico, prevención o tratamiento de una enfermedad rara- para una dolencia mitocondrial rara y grave.
Un estudio pionero de la Universidad de Granada (UGR), en el sur de España, y el Hospital Universitario de Düsseldorf (Alemania) ha abierto una nueva vía terapéutica para enfermedades metabólicas pediátricas y ha permitido desarrollar un medicamento huérfano -destinado al diagnóstico, prevención o tratamiento de una enfermedad rara- para una dolencia mitocondrial rara y grave.
Investigadores de la UGR y del Hospital Universitario de Düsseldorf han liderado un estudio pionero, publicado en la revista Brain, que demuestra por primera vez la eficacia de un tratamiento racionalmente diseñado para la deficiencia primaria de coenzima Q (CoQ), una enfermedad mitocondrial rara y grave.
La publicación se produce, precisamente, en los días previos a la celebración de la semana internacional de las enfermedades mitocondriales, programada del 15 al 19 de septiembre.
La investigación se ha basado en un modelo con roedores generado en la UGR que reproduce fielmente la enfermedad humana causada por mutaciones en el gen COQ2 y que ha permitido estudiar en detalle los mecanismos fisiopatológicos y evaluar la eficacia terapéutica de un compuesto fenólico.
Los resultados preclínicos demostraron que la administración crónica del compuesto prevenía la encefalopatía mitocondrial y mejoraba la supervivencia, sentando así las bases para una traslación racional y segura al paciente.
El paso a la práctica clínica se ha realizado en un niño de tres años con un cuadro clínico muy grave, un síndrome nefrótico resistente a esteroides y encefalopatía con lesiones cerebrales tipo Leigh, con un pronóstico muy desfavorable.
El tratamiento con el compuesto fenólico ha conseguido una remisión completa de la nefropatía y una mejoría significativa del cuadro neurológico y que, en unos seis meses, el niño que no podía caminar ni interactuar con normalidad haya empezado a andar de forma independiente.
Además, ha recuperado un 20% de su peso corporal y ha mostrado una clara mejoría en el lenguaje, la interacción social y la capacidad cognitiva.
«Ha sido muy emocionante comprobar que un tratamiento en el que hemos trabajado durante años en el laboratorio haya podido cambiar la vida de un niño y la de su familia. Es una recompensa como investigadora», ha explicado Julia Corral Sarasa, primera autora del estudio.
Estos resultados no solo ofrecen una esperanza real para pacientes sin opciones terapéuticas, sino que también tienen un claro potencial de transferencia y aplicación clínica.
La Universidad de Granada es la institución que ha solicitado la designación de medicamento huérfano de la Agencia Europea del Medicamento y la patente internacional que protege esta estrategia terapéutica.
Además, la iniciativa cuenta con el apoyo de la Oficina de Transferencia de Resultados de Investigación (OTRI-UGR) para avanzar en su valorización con el fin de alcanzar impacto sanitario y socioeconómico, por lo que se exploran otras aplicaciones médicas.
15 septiembre 2025 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jul
10
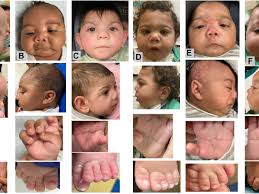 La Organización Panamericana de la Salud (OPS) presentó hoy una plataforma interactiva que consolida datos en tiempo real sobre anomalías congénitas reportadas por los sistemas nacionales de vigilancia epidemiológica de América Latina y el Caribe.
La Organización Panamericana de la Salud (OPS) presentó hoy una plataforma interactiva que consolida datos en tiempo real sobre anomalías congénitas reportadas por los sistemas nacionales de vigilancia epidemiológica de América Latina y el Caribe.
Llamada Repositorio de Defectos Congénitos en las Américas, esta herramienta busca fortalecer la toma de decisiones basadas en evidencia, apoyando la formulación de políticas y estrategias para mejorar la salud materno-infantil.
Datos del organismo sanitario muestran que en la región nacen cada año unos 15 millones de niños, 10 millones de ellos en América Latina y el Caribe.
Más de la mitad de las muertes en menores de cinco años ocurre en el primer mes de vida, principalmente por prematuridad, defectos congénitos, sepsis o asfixia.
Las anomalías al nacer son una de las principales causas de mortalidad neonatal y discapacidad infantil, afectando a miles de familias.
Según los expertos, estas condiciones están asociadas a factores genéticos y hereditarios, anomalías cromosómicas, exposiciones ambientales, deficiencias nutricionales, exposición a sustancias tóxicas o enfermedades crónicas o infecciosas durante el embarazo.
En respuesta a esta situación, el repositorio ofrece paneles interactivos que muestran tendencias, distribución geográfica y patrones demográficos de los defectos congénitos, facilitando el diseño de estrategias de prevención e intervenciones sanitarias.
Aunque la plataforma presenta en estos momentos datos preliminares de algunos países, su cobertura se ampliará a medida que más autoridades nacionales incorporen sus registros, con el apoyo técnico de la OPS.
Actualmente, 14 países de la región cuentan con programas nacionales de vigilancia de anomalías congénitas, en distintos niveles de desarrollo.
Durante el seminario virtual de presentación de la nueva herramienta, cuatro de ellos (Argentina, Brasil, Costa Rica y Cuba) compartieron sus experiencias, así como los desafíos clave en la implementación de estos sistemas.
La OPS invitó a los países de la región a unirse a esta plataforma colaborativa, fortaleciendo la vigilancia epidemiológica y avanzando hacia un futuro donde cada niño tenga la oportunidad de un comienzo saludable.
25 junio 2025 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jun
23
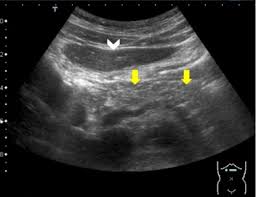 La Sociedad Española de Patología Digestiva (SEPD) señala que la esteatosis pancreática se trata de una entidad relativamente reciente y emergente que, pese a estar considerada como una entidad independiente y con características específicas que la distinguen de otras enfermedades, los mecanismos etiopatogénicos que conducen a su desarrollo, así como las complicaciones que supone a largo plazo, están aún poco caracterizadas.
La Sociedad Española de Patología Digestiva (SEPD) señala que la esteatosis pancreática se trata de una entidad relativamente reciente y emergente que, pese a estar considerada como una entidad independiente y con características específicas que la distinguen de otras enfermedades, los mecanismos etiopatogénicos que conducen a su desarrollo, así como las complicaciones que supone a largo plazo, están aún poco caracterizadas.
Este ha sido el punto principal que se ha desarrollado en la ponencia ‘Esteatosis pancreática: desafíos actuales y perspectivas futuras’, de la Mesa SEPD / AESPANC que ha tenido lugar durante la celebración del 84º Congreso de la SEPD en Bilbao.
En la ponencia, la experta de la SEPD y especialista en aparato digestivo del Hospital Universitario Costa del Sol, Cristina Verdejo, ha indicado que uno de los mayores desafíos que enfrenta la comunidad médica en relación con esta condición es la falta de estandarización en los métodos diagnósticos, en los criterios de valores de corte, y en las pautas de seguimiento establecidas.
«La esteatosis pancreática es una condición que, pese a su creciente prevalencia, no cuenta aún con una definición clara y consensuada a nivel internacional. Existen diferencias terminológicas, y no está presente en la Clasificación Internacional de Enfermedades (CIE), por ejemplo», subraya la experta.
Al hilo, Verdejo ha destacado que es una entidad que ha desatado un creciente interés en los últimos años debido a que «conlleva una serie de implicaciones que contribuyen a causar o agravar procesos inflamatorios, metabólicos, neoplásicos e, incluso, cáncer de páncreas». Entre estos procesos destacan la disfunción pancreática endocrina y exocrina, pancreatitis aguda, pancreatitis crónica, neoplasia pancreática o un mayor riesgo de fistula pancreática postoperatoria (FPPO).
Actualmente, «existen herramientas invasivas y no invasivas para evaluar los depósitos de grasa intrapancreáticos», indica Verdejo, al tiempo que añade que algunas de estas herramientas cuentan con una alta precisión, como son la resonancia magnética, la tomografía computarizada y la ecoendoscopia. Sin embargo, «la evaluación de los depósitos de grasa intrapancreática requiere de un método de examen estandarizado con valores de corte significativos y validados prospectivamente que, hoy en día, no tenemos», confirma.
«La evaluación de los depósitos de grasa intrapancreática podría convertirse en un futuro, por ejemplo, como marcador temprano de la resistencia a la insulina e identificar a pacientes con riesgo de diabetes que, actualmente, no son detectados por los enfoques convencionales. Así como en personas que padecen síndrome metabólico, este se puede tomar como marcador pronóstico de insuficiencia pancreática exocrina, de complicaciones postoperatorias como la FPPO, de pancreatitis crónica y/o de cáncer de páncreas», asegura la experta.
En este punto, la especialista ha dicho que actualmente no existen directrices de consenso para su tratamiento, así como que la investigación sobre la eficacia de fármacos está limitada a estudios preclínicos y clínicos a pequeña escala y se necesita investigación clínica más fiable para explorar y validar su eficacia.
Actualmente, las modificaciones en el estilo de vida, como la adopción de una dieta equilibrada, el ejercicio regular y el abandono del hábito tabáquico, son fundamentales para la gestión de la enfermedad. «El tratamiento de la EP se centra en la prevención, controlando los factores de riesgo y adoptando un enfoque integral», añade.
PERSPECTIVAS FUTURAS
De cara al futuro, se espera que la investigación sobre la esteatosis pancreática «avance considerablemente, con estudios prospectivos que profundicen en su comprensión y en la identificación de mecanismos etiopatogénicos aún poco claros», declara la experta.
«La estandarización de métodos diagnósticos y la definición de criterios de corte clínicos serán esenciales para mejorar la precisión en el diagnóstico y el manejo de esta entidad», indica Verdejo, que destaca que se prevé el desarrollo de pautas clínicas que permitan una caracterización integral de los depósitos de grasa intrapancreática, lo que facilitaría su integración en el tratamiento rutinario, de manera similar a la evaluación de la grasa hepática.
Por último, la SEPD ha explicado que la esteatosis pancreática es una infiltración de tejido graso en las células del páncreas que puede ser local, afectando solo a una parte del páncreas, o difusa afectando a la totalidad del órgano.
En este sentido, apunta, la penetración difusa tiende a tener un mayor impacto sistémico y puede asociarse a complicaciones metabólicas y neoplásicas más graves, mientras que la penetración localizada puede estar vinculada a condiciones más específicas y focalizadas. Con esto, el grado de infiltración de grasa afecta considerablemente a las implicaciones clínicas que conllevan. Cuando se produce el reemplazo graso, se cree que ya es irreversible, debido a la muerte de las células acinares pancreáticas.
16 mayo 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jun
17
 Según la evidencia científica, los pacientes con patología dual tienen una elevada incidencia de insomnio, que se mueve en una horquilla de entre el 50% y el 80%, dependiendo de la sustancia principal de adicción. Además, el insomnio también se asocia a mayor probabilidad de recaídas en el consumo y de descompensaciones psiquiátricas.
Según la evidencia científica, los pacientes con patología dual tienen una elevada incidencia de insomnio, que se mueve en una horquilla de entre el 50% y el 80%, dependiendo de la sustancia principal de adicción. Además, el insomnio también se asocia a mayor probabilidad de recaídas en el consumo y de descompensaciones psiquiátricas.
Por un lado, el insomnio predispone a las adicciones y a otras enfermedades de salud mental, provocando el desarrollo de patología dual. Por otro, la mayoría de las afecciones psiquiátricas se asocian a insomnio.
En este sentido, la psiquiatra ha explicado que «este círculo se puede romper favoreciendo la abstinencia al consumo de sustancias, y realizando un tratamiento adecuado tanto para las patologías psiquiátricas como para el trastorno por insomnio».
Para ello, la doctora ha afirmado que la evidencia científica indica que el tratamiento más eficaz para el insomnio en todas las enfermedades es la psicoterapia cognitivo-conductual. Sin embargo, matizó que, en pacientes duales que presentan mayor gravedad, a esta terapia se debe sumar en muchas ocasiones el uso de fármacos.
«En la actualidad los antagonistas duales de los receptores de la orexina (DORA) son una alternativa terapéutica muy eficaz para el insomnio», ha asegurado Grau. Las orexinas son unas moléculas producidas por unos miles de neuronas en el hipotálamo que son fundamentales para la estabilidad del ciclo vigilia/sueño.
En esta línea, el profesor de Psiquiatría de la Universidad de Stanford, Luis de Lecea, ha explicado que «en el insomnio el cerebro no acaba de procesar bien cuándo necesita dormir. Si bloqueamos la actividad de las neuronas que producen hipocretina aumentamos la probabilidad de que los circuitos cerebrales procesen bien esa información».
Según el experto, esto también puede tener un impacto sobre la salud mental. «La falta de sueño está relacionada con prácticamente todos los trastornos neuropsiquiátricos. Por ello, los antagonistas de receptores de la orexina son buenos candidatos a reducir la actividad dopaminérgica, que suele estar elevada en trastornos de ansiedad y abuso de sustancias», ha señalado de Lecea.
Por último, el experto ha destacado que los DORA son mucho más efectivos para el manejo del insomnio que los fármacos Z o las benzodiacepinas. «Estos fármacos son bastante efectivos para reducir la latencia del sueño, pero son muy inespecíficos; actúan sobre receptores cerebrales que inhiben la transmisión sináptica en muchas partes del cerebro. Los antagonistas de receptores de orexina, en cambio, son bastante más selectivos en cuanto a dónde actúan», concluye.
09 junio 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jun
9
 Familiares de fallecidos en residencias madrileñas durante la pandemia han clamado este domingo contra los «protocolos de la vergüenza» de la Comunidad de Madrid, y han pedido al Gobierno central que constituya una comisión de investigación para «saber lo que pasó y para sacar lecciones» de lo ocurrido.
Familiares de fallecidos en residencias madrileñas durante la pandemia han clamado este domingo contra los «protocolos de la vergüenza» de la Comunidad de Madrid, y han pedido al Gobierno central que constituya una comisión de investigación para «saber lo que pasó y para sacar lecciones» de lo ocurrido.
Bajo el lema «Verdad, Justicia y Reparación», los asistentes, convocados por las asociaciones 7 291 Verdad y Justicia y Pladigmare, han solicitado además al Ejecutivo regional y al PP que se reabra la comisión de investigación de la Asamblea de Madrid con la participación de familiares y trabajadoras de las residencias.
«Estamos aquí para rendir homenaje a las 7 291 personas que se murieron de una manera indigna y cruel por unos protocolos y una decisión política que los llevó directamente a esa muerte», ha explicado María Jesús Valero, de 7 291 Verdad y Justicia, antes del acto al que han asistido representantes del PSOE y Más Madrid.
En un comunicado, la asociación denuncia la «decisión política» del Gobierno de la Comunidad de Madrid, que acordó «que las personas que vivían en residencias y eran dependientes o sufrían deterioro cognitivo se quedasen sin asistencia sanitaria».
En la misma línea se expresa Pladigmare, que lamenta que dicha postura en los primeros meses de la pandemia costara la vida, «de forma indigna, a 7 291 personas».
«No hablamos desde el rencor, sino desde la necesidad de memoria, justicia y acción», añade la asociación, que defiende que «el tiempo no borra el daño cuando no hay reparación, y el silencio institucional no cura: solo alimenta el olvido y perpetúa el maltrato».
Desde su punto de vista, las decisiones políticas «tomadas sin previsión ni humanidad se convirtieron en una forma de maltrato institucional», lo cual «deshumanizó a las víctimas y se relegó su dignidad».
Aunque los familiares sienten que el daño «ha sido profundo, sostenido» y «sigue vivo en cuerpos, hogares y corazones», reconocen que «hay motivos para la esperanza», sobre todo a raíz de la apertura de varias causas judiciales, la última por un juzgado de Leganés, contra varios ex altos cargos del Gobierno regional por la gestión de las residencias durante la primera ola de la covid.
«Trabajar por el cuidado y la dignidad de los mayores es el mejor homenaje que podemos hacer a la pérdida y el recuero digno de esas 7 291 personas», concluyen desde Pladigmare.
Un acto con presencia de ministros
Hasta la céntrica plaza madrileña en la que ha transcurrido el acto se han acercado el secretario general del PSOE-M y ministro para la Transformación Digital y de la Función Pública, Óscar López, y las portavoces de Más Madrid, Manuela Bergerot y Rita Maestre.
Todos han insistido en que la justicia dará la «verdad» a los familiares de las víctimas del coronavirus de los conocidos como ‘protocolos de la vergüenza’ que impidieron la derivación de personas mayores de residencias a hospitales al comienzo de la crisis sanitaria.
También ha acudido la ministra de Sanidad, Mónica García, quien considera que la presidenta de la Comunidad de Madrid, Isabel Díaz Ayuso, debería «dejar de insultar, dejar de acosar y de hablar del Gobierno de España» y, en su lugar, sentarse con los familiares de los fallecidos en las residencias madrileñas durante la pandemia.
Homenaje a los fallecidos en la dana
Los asistentes han homenajeado, asimismo, a los más de 200 fallecidos, en su mayoría, en la provincia de Valencia, durante la dana del pasado 29 de octubre.
Para 7 291 Verdad y Justicia, «hay similitudes» entre los muertos en las residencias madrileñas en la pandemia y las personas que fallecieron en la Comunidad Valenciana durante la dana, como son «la negligencia y, sobre todo, que (ambas regiones) intentan ocultar la verdad».
01 junio 2025 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
jun
6
 La Federación Española de Enfermedades Raras (FEDER), junto con hospitales de la red Únicas y la compañía Alexion, han lanzado una campaña para mejorar la investigación y la capacidad de diagnóstico de las enfermedades raras pediátricas.
La Federación Española de Enfermedades Raras (FEDER), junto con hospitales de la red Únicas y la compañía Alexion, han lanzado una campaña para mejorar la investigación y la capacidad de diagnóstico de las enfermedades raras pediátricas.
Según ha explicado este martes el presidente de la FEDER, Juan Carrión, en un comunicado, «los niños con enfermedades raras tardan una media de seis años en recibir un diagnóstico», y ha añadido que el 20% de ellos han tenido que esperar más de 10 años para obtenerlo.
Ante este reto, en 2021 se creó el proyecto Únicas, una red de 30 hospitales españoles liderados por el Ministerio de Sanidad que tienen como una de sus prioridades aumentar el número de niños con enfermedades raras sin diagnóstico que se sometan a estudios genéticos avanzados.
Estos pacientes se derivan al proyecto IMPaCT-GENóMICA, impulsado por el Instituto de Salud Carlos III y el CIBER de Enfermedades Raras (CIBERER), donde se les hace un estudio genético completo y un grupo de expertos analiza los resultados con el objetivo de encontrar variantes genéticas que puedan determinar qué enfermedad padecen.
«El diagnóstico no lo es todo, pero lo cambia todo», ha afirmado la directora estratégica del proyecto Únicas del Hospital Sant Joan de Déu, Encarna Guillén, que ha remarcado que «sin diagnóstico no hay pronóstico» y, por tanto, «no se sabe cómo evolucionará la enfermedad ni es posible abordar la causa y tratarla».
Además, Guillén ha lamentado que, sin la diagnosis tampoco pueden conocer el método de transmisión de la enfermedad y no pueden «prevenir otros casos en la familia mediante asesoramiento genético».
Según datos de la FEDER, se estima que el 80% de las enfermedades raras detectadas son de base genética.
Para poder financiar el proyecto Únicas, FEDER, el Hospital Sant Joan de Déu de Barcelona y otros centros de la red Únicas, y Alexion -la división de enfermedades raras de la farmacéutica AstraZeneca- han iniciado este martes, coincidiendo con el Día Nacional del Niño Hospitalizado, la campaña ‘El Viaje de Nica’, que pretende sensibilizar a la sociedad sobre esta realidad y captar fondos.
De este modo, un centenar de familias con niños afectados por enfermedades raras pondrán en circulación 100 muñecas de Nica, que viajarán de mano en mano para recrear el periplo que muchas familias con niños afectados por este tipo de enfermedades tienen que hacer para recibir un diagnóstico.
Las muñecas tendrán un código QR que da acceso a una web donde la familia que empezó la cadena explica su caso particular y donde se podrá realizar una donación monetaria a la causa.
«Es la forma en que muchas familias con niños y niñas con enfermedades raras dejen de ser invisibles», han asegurado los padres de una niña con acidura metilmalónica con homocistinuria, Toni y Maria, que han añadido que este tipo de iniciativas les dan esperanza.
13 mayo 2025 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
