
abr
21
 La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
La enfermedad de Pompe presentada por primera vez en 1930 sigue siendo hoy uno de los males que afecta la calidad de vida de los seres humanos sin distinguir entre edad y sexo.
De acuerdo con la literatura médica, es considerada poco frecuente y que aqueja a hombres y mujeres de cualquier edad, sin importar el origen étnico, e incluso puede originarse en cualquier etapa de vida del paciente desde lactantes hasta adultos.
Es una enfermedad neuromuscular hereditaria autosómica recesiva y de origen genético, causada por un gen que genera el déficit de la enzima alfa-glucosidasa ácida o maltasa ácida.
Ello permite la acumulación de una sustancia en los músculos denominada glucógeno, ocasionando pérdida de masa muscular, así como debilidad muscular en las extremidades inferiores.
Igualmente puede afectar los músculos del tronco, brazos y hombros, y la gravedad y síntomas varían en cada paciente.
Entre los malestares relacionados con esta dolencia genética están la dificultad para levantarse, al estar sentado, para caminar, subir escaleras, masticar y la debilidad muscular progresiva.
Una persona aquejada por la Enfermedad de Pompe, también puede presentar reflujo gastroesofágico, disnea, caídas frecuentes, agitación tras realizar esfuerzo físico, dolor de cabeza por las mañanas y somnolencia durante el día debido a la ortopnea.
El nombre de esta dolencia es en honor al patólogo holandés Joannes Pompe, quien presentó el primer caso en el año 1930, y cada 15 de abril es celebrado el Día Internacional de la Enfermedad de Pompe.
15 abril 2026 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
mar
25
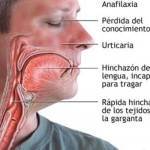 Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
Investigadores del Hospital Universitario Vall d’Hebron de Barcelona y del Hospital Universitario de Santa María de Lleida han identificado el primer caso documentado en el mundo de transmisión de una enfermedad minoritaria -angioedema hereditario- a través de un donante de esperma anónimo en una clínica de reproducción asistida.
El trabajo, publicado en la revista científica Frontiers in Immunology, destaca la identificación de varios casos portadores de la misma variante genética (T328K gen F12), todos ellos concebidos mediante reproducción asistida a través de un único donante.
Angioedema hereditario
El angioedema hereditario es una enfermedad genética muy poco frecuente que provoca hinchazón en la piel, aparato digestivo o vías respiratorias; unos síntomas que afectan al 60-80 % de las mujeres portadoras, frente a una probabilidad muy baja en los hombres.
La investigación arrancó con una paciente que presentaba episodios recurrentes de hinchazón en la cara, sin responder a los tratamientos antialérgicos habituales, y cuyo análisis genético identificó la variante T328K.
Esta fue también detectada posteriormente en otras personas concebidas con el esperma del mismo donante, mientras que no estaba presente en la línea materna analizada.
Ante la sospecha de que la mutación genética podía proceder del padre, los investigadores contactaron con la clínica de fertilidad y, tras coordinar un estudio con el banco de esperma, pudieron confirmar que este era portador heterocigoto de la variante.
Notificación de la clínica
La clínica notificó entonces a las mujeres que habían utilizado el esperma de dicho donante, un procedimiento que permitió identificar a más portadores, algunos de ellos sin manifestaciones sintomáticas.
«Desde el punto de vista genético, este caso es especialmente relevante porque demuestra cómo una variante patogénica puede pasar desapercibida en un donante asintomático y transmitirse a varios descendientes», explica el jefe del grupo de Inmunología Traslacional del Vall d’Hebron Institut de Recerca (VHIR), Roger Colobran.
Los investigadores subrayan que este caso pone de relieve los retos que plantean las enfermedades genéticas hereditarias asintomáticas en los programas de donación de gametos, ya que el cribado genético de los donantes se centra principalmente en enfermedades infecciosas.
En este sentido, señalan que, en regiones donde esta variante es más prevalente, podría valorarse el cribado dirigido al gen F12.
20 marzo 2026 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2026. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
oct
8
 De este modo, la Sociedad Balear de Laboratorio Clínico, Asociación Canaria del Laboratorio Clínico (ACLAC), Asociación de Analistas Clínicos de Extremadura (ANCLEX), Asociación Valenciana de Especialistas en Laboratorio Clínico (AVELAC), Sociedad Castellanomanchega de Medicina de Laboratorio (LABCAM), Sociedad Asturiana de Bioquímica y Análisis Clínicos – Medicina de Laboratorio (SAByAC ML), Sociedad Andaluza de Análisis Clínicos, Medicina del Laboratorio (SANAC) y Sociedad Castellanoleonesa de Análisis Clínicos (SCLAC) y la Sociedad de Medicina de Laboratorio de la región de Murcia (SOMELMUR), apuestan por la creación de un Diploma de Acreditación Avanzada para esta área funcional que consideran «constituye una alternativa viable».
De este modo, la Sociedad Balear de Laboratorio Clínico, Asociación Canaria del Laboratorio Clínico (ACLAC), Asociación de Analistas Clínicos de Extremadura (ANCLEX), Asociación Valenciana de Especialistas en Laboratorio Clínico (AVELAC), Sociedad Castellanomanchega de Medicina de Laboratorio (LABCAM), Sociedad Asturiana de Bioquímica y Análisis Clínicos – Medicina de Laboratorio (SAByAC ML), Sociedad Andaluza de Análisis Clínicos, Medicina del Laboratorio (SANAC) y Sociedad Castellanoleonesa de Análisis Clínicos (SCLAC) y la Sociedad de Medicina de Laboratorio de la región de Murcia (SOMELMUR), apuestan por la creación de un Diploma de Acreditación Avanzada para esta área funcional que consideran «constituye una alternativa viable».
La genética de laboratorio forma parte del núcleo competencial del Laboratorio Clínico y ha sido desarrollada durante décadas con altos estándares de calidad, responsabilidad y evidencia científica. «No se trata de una disciplina externa ni añadida, sino de una competencia estructural inherente que los especialistas ejercen con solvencia y rigor, respaldados por su trayectoria y resultados clínicos contrastados», afirman.
Las entidades firmantes consideran que cualquier revisión del mapa de especialidades sanitarias debe fundamentarse en un análisis «exhaustivo y riguroso» que evalúe sus repercusiones sobre el Laboratorio Clínico, garantizando así la preservación de la excelencia asistencial. En este sentido, apelan a la «serenidad, la cohesión y la claridad» entre los profesionales, promoviendo un diálogo constructivo sustentado en una formación sanitaria especializada en genética diagnóstica, biología molecular y medicina de precisión, pilares fundamentales para la calidad asistencial y la seguridad del paciente.
Consideran que corresponde a la Comisión Nacional de la Especialidad actualizar los programas formativos y proponer, cuando sea necesario, medidas de refuerzo utilizando las herramientas oficiales disponibles, como la ampliación del periodo formativo a 5 años o la creación de Áreas de Capacitación Específica, conforme a lo establecido en el Real Decreto 589/2022.
Las sociedades científicas consideran que la propuesta del Ministerio «no responde a carencias formativas o tecnológicas reales, sino a una problemática laboral generada por la contratación de personal no sanitario sin la formación sanitaria especializada requerida para el desempeño de funciones asistenciales». Según su experiencia, la solución no debe centrarse en la fragmentación de competencias, sino en el fortalecimiento y consolidación de las estructuras formativas actualmente vigentes.
Además, advierten que esta propuesta vulnera los principios establecidos en el Real Decreto 589/2022, normativa que dirige cualquier regulación hacia la eficiencia, la cohesión y la calidad asistencial.
«La medida no solo impide una gestión racional de los recursos públicos, sino que también fomenta la fragmentación de la formación de los profesionales y de la asistencia sanitaria, generando compartimentos estancos que comprometen la continuidad y la integración de los cuidados», afirman.
No obstante, las entidades reiteran su firme disposición a colaborar con la Administración sanitaria en el diseño de alternativas fundamentadas en la eficiencia, la evidencia científica y la coherencia estructural. Confían en que el trabajo conjunto permitirá adecuar «la oferta formativa a las necesidades reales del sistema sanitario y de la ciudadanía, evitando duplicidades y solapamientos contraproducentes».
29 septiembre 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
oct
2
 Aunque la mayoría de los casos de cáncer nace de mutaciones espontáneas o factores ambientales, cerca de una cuarta parte tiene un componente hereditario, por eso, hacer cribados genéticos para estudiar la predisposición genética de los pacientes, en vez de realizar pruebas como mamografías o colonoscopias, salvaría muchas vidas.
Aunque la mayoría de los casos de cáncer nace de mutaciones espontáneas o factores ambientales, cerca de una cuarta parte tiene un componente hereditario, por eso, hacer cribados genéticos para estudiar la predisposición genética de los pacientes, en vez de realizar pruebas como mamografías o colonoscopias, salvaría muchas vidas.
Porque «si nos anticipamos, podemos avanzar mucho más» en la lucha contra el cáncer, ha subrayado el catedrático de Medicina Legal de la Universidad de Santiago de Compostela y coordinador del proyecto IMPACT del Instituto de Salud Carlos III, Ángel Carracedo.
Mutaciones y cáncer hereditario
Carracedo ha hecho estas afirmaciones en su intervención en una jornada organizada por el Centro Nacional de Investigaciones Oncológicas (CNIO) para conmemorar el Día Mundial de la Investigación del Cáncer, donde ha explicado que las mutaciones genéticas son esenciales para la vida y para la diversidad, pero lamentablemente «a veces pueden producir enfermedades como el cáncer».
Algunas de estas mutaciones se conocen y son responsables de entre un 5 y un 10% de los casos de cáncer pero otras son hereditarias y se agrupan en lo que se denomina «síndrome de cáncer hereditario», es decir, trastornos genéticos que aumentan el riesgo de padecer ciertos tipos de cáncer en una misma familia.
Este síndrome es responsable de entre un 15 y un 20% de los cánceres familiares, pero «queda mucho por descubrir», reconoce el genetista.
Señales de cáncer familiar
«Existen varias señales que pueden indicar que estamos ante un cáncer familiar, como la aparición de tumores en niños y en menores de 20 años, el cáncer bilateral (se produce en órganos pares -ojos, riñones, pulmones-…), cuando hay más de un tipo de cáncer en una sola persona o cuando un cáncer se repite en muchas generaciones», ha explicado Mercedes Robledo, del grupo de Cáncer Endocrino Hereditario del CNIO e investigadora del programa IMPACT.
Ayudar a las familias a las que ha atacado el cáncer es el objetivo principal «del programa IMPACT, que tiene más de un millar de especialistas dedicados a buscar respuestas para estas personas», ha explicado Robledo.
El programa, dividido en varios ejes (Cohorte, Data y Genómico) pretende, entre otras cuestiones, servir para identificar la causa genética en las familias en las que hay cáncer, pero no saben qué mutación o gen las causa y, para ello, se están recogiendo datos de 20 000 personas en España para estudiar los factores genéticos del cáncer.
Un proyecto para secuenciar 20 000 genomas
A largo plazo, el objetivo IMPACT es confeccionar una cohorte de 200 000 personas formada por personas de todo el país, de entre 19 y 79 años, a los que se hará un seguimiento de salud global de 20 años, ha explicado la vicedirectora del Instituto de Salud Carlos III (ISCIII), Rosario Perona.
De ellos, se recogerán muestras y se secuenciarán unos 20 000 genomas -ha dicho-, que serán una imagen de referencia del «genoma español» que permitirá saber cómo es la salud de los españoles y que, además, podrá ser utilizada por los investigadores tanto para estudios científicos y medicina personalizada, y por supuesto para avanzar en el conocimiento del cáncer.
Por su parte, la jefa de la unidad de Cáncer Familiar del CNIO, María Currás, ha explicado que, desde su consulta en el Hospital de Fuenlabrada, estudia más de 600 casos al año en los que intenta averiguar si hay un cáncer familiar: «El 30% de mis pacientes son personas sanas con un familiar con cáncer que sospechan que tienen un tumor familiar».
Para llegar a una consulta como la suya, hay que solicitar al médico de familia que traslade su caso al consejo genético de su hospital de referencia, donde los especialistas en genética intentan averiguar si el paciente es portador de una mutación o no porque «la información es poder», ha subrayado la doctora del CNIO.
Si las personas sospecharan que tienen una mutación causante de cáncer, en muchos casos podría detectarse con un análisis de sangre, y tomar medidas preventivas adecuadas, como extirparse un órgano o someterse a revisiones periódicas para detectar el cáncer a tiempo, ha detallado.
23 septiembre 2025 | Fuente: EFE | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
may
8
 En la Franja de Gaza aumentó significativamente el número de casos de malformaciones fetales y partos complicados debido al uso de armas prohibidas por parte del Ejército israelí, afirmó hoy la agencia de noticias palestina Shehab.
En la Franja de Gaza aumentó significativamente el número de casos de malformaciones fetales y partos complicados debido al uso de armas prohibidas por parte del Ejército israelí, afirmó hoy la agencia de noticias palestina Shehab.
Solo en junio, julio y agosto de 2024 nacieron allí 172 bebés con deformidades y trastornos congénitos, destacó la fuente, aunque aclaró que es imposible conocer la cifra exacta por la destrucción causada al sistema de salud.
«La tragedia es acumulativa en sus datos y consecuencias: ni siquiera los fetos en el vientre de sus madres se salvaron del exterminio», denunció el medio de prensa.
Destacó que decenas de fetos nacieron o fueron abortados con cuerpos deformados.
«En Gaza, la deformidad no es una rara coincidencia médica, sino un indicador inquietante de una catástrofe humanitaria silenciosa que se propaga a través de las generaciones venideras», alertó.
La fuente citó informes médicos y testimonios, según los cuales, hay un aumento significativo de esos casos, motivado por la utilización de armas prohibidas internacionalmente.
Mientras que los hospitales de la sitiada Franja carecen del equipamiento médico necesario para un diagnóstico y tratamiento tempranos, los residentes se ven obligados a valerse por sí mismos, indicó.
Shehab criticó el «vergonzoso silencio internacional y el desprecio por los derechos humanos básicos».
Como ejemplo se refirió a los casos de Malak Ahmed Al-Qanou, una niña que nació sin cerebro en el norteño hospital Al-Awda, y Muhammad Abu Awad, quien tiene deformidades congénitas en el corazón, los pies y el cerebro.
También documentó el caso de Noura Abu Marouf, una bebé que nació muerta debido a defectos cerebrales congénitos y un agujero en el corazón.
Munir al-Barash, director general del Ministerio de Salud en Gaza, destacó que los médicos palestinos atribuyen el problema al uso por parte de Israel de armas experimentales con efectos radiológicos y químicos devastadores.
Al-Barash señaló la similitud entre lo que sucede hoy en Gaza y lo que ocurrió en Iraq después de la invasión estadounidense, cuando se registró un aumento sin precedentes de defectos de nacimiento debido a la contaminación y la radiación.
Por ello reclamó una investigación internacional urgente sobre la naturaleza de las armas utilizadas por el Ejército israelí.
En similar sentido se pronunció el doctor Hatem Zaheer, jefe del departamento de enfermería del Complejo Médico Nasser.
Zaher afirmó que el aumento del número de recién nacidos que sufren malformaciones congénitas, en particular defectos cardíacos, reproductivos y de las extremidades inferiores está motivada por «la guerra de exterminio en la que se lanzaron millones de materiales explosivos sobre la Franja de Gaza».
05 mayo 2025 | Fuente: Prensa Latina | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
abr
22
 Este descubrimiento ofrece «respuestas» y «esperanza» a miles de familias que llevan años buscando el diagnóstico de una serie de trastornos de base genética que se manifiestan en la primera infancia, y que pueden provocar situaciones altamente discapacitantes.
Este descubrimiento ofrece «respuestas» y «esperanza» a miles de familias que llevan años buscando el diagnóstico de una serie de trastornos de base genética que se manifiestan en la primera infancia, y que pueden provocar situaciones altamente discapacitantes.
«Sabemos por años de experiencia en el apoyo a pacientes y familias con enfermedades genéticas raras que recibir un diagnóstico como este puede cambiar la vida y ser el primer paso para poner en marcha el apoyo y la atención adecuados», ha destacado el coordinador de la infraestructura IMPaCT-Genómica, Ángel Carracedo, que ha participado en la investigación junto con los programas de enfermedades raras no diagnosticadas ENoD-CIBERER y URDcat.
Tras ello, ha resaltado que la red de hospitales participantes en la infraestructura IMPaCT-Genómica y otros programas de enfermedades no diagnosticadas pueden aprovecharse de estos «punteros» descubrimientos y mejorar «rápidamente» las tasas de diagnóstico a nivel local.
Los científicos han señalado que los avances en las técnicas de secuenciación del genoma completo y la exploración de regiones no codificantes en grandes cohortes de pacientes han supuesto reciente y gran avance en la genética de los trastornos del neurodesarrollo.
«Trabajar con datos genómicos de cohortes grandes de pacientes bien caracterizados fenotípicamente, con sistemas de análisis masivos y de intercambio de datos, brinda la oportunidad de acelerar el descubrimiento de nuevos hallazgos y validarlos rápidamente, como se demuestra en este estudio en el que colaboramos», ha señalado Carracedo.
El equipo de investigadores, a partir del descubrimiento del síndrome RNU4-2/ReNU el año pasado, uno de los tipos monogénicos más comunes de los trastornos del neurodesarrollo, ha logrado identificar este nuevo síndrome relacionado que está causado por mutaciones en el gen no codificante RNU2-2.
Tras analizar los datos de miles de personas con trastornos del neurodesarrollo se han podido hallar nueve casos con mutaciones de novo en este gen, a los que se han sumado otros 16 casos fruto del análisis masivo de otras ocho colecciones de enfermedades raras no diagnosticadas.
La identificación de las mutaciones en RNU2-2 como nueva causa de los trastornos del neurodesarrollo es «especialmente notable» debido a que consolida la importancia biológica de estos genes en ese tipo de afecciones, pues tienden a producirse espontáneamente en lugar de heredarse de los progenitores.
Por su parte, la colaboradora de la coordinación de ENoD-CIBERER e IMPaCT-Genómica, Beatriz Morte, ha expresado que este descubrimiento también posibilita nuevas investigaciones encaminadas a explorar los mecanismos moleculares subyacentes al trastorno.
La experta también ha explicado que los síndromes RNU4-2/ReNU y RNU2-2 comparten similitudes, pero que los pacientes con síndrome RNU2-2 suelen estar más afectados por la epilepsia.
«Se estima que la prevalencia del trastorno RNU2-2 es aproximadamente el 20 % de la del síndrome RNU4-2/ReNU, uno de los tipos monogénicos más comunes de los trastornos del neurodesarrollo. Esto significa que debe haber miles de familias afectadas en todo el mundo», ha agregado.
Esta investigación ha servido igualmente para identificar una mutación distinta en RNU2-2 que tiende a aparecer en individuos no afectados a medida que envejecen, lo que puede tener implicaciones para las afecciones relacionadas con la edad.
Los programas españoles involucrados en el estudio son IMPaCT-Genómica, con cerca de 2 000 pacientes que provienen de todas las comunidades autónomas, y que es una iniciativa del Instituto de Salud Carlos III gestionada por el Centro de Investigación Biomédica en Red (CIBER), el Programa de Enfermedades no Diagnosticadas del CIBER de Enfermedades Raras (ENoD-CIBERER) y el programa de enfermedades no diagnosticadas de la Generalitat de Catalunya (URDCat). El estudio también ha contado con la colaboración de equipos de Reino Unido, Bélgica, Países Bajos e Islandia.
15 abril 2025 | Fuente: Europa Press | Tomado de la Selección Temática sobre Medicina de Prensa Latina. Copyright 2025. Agencia Informativa Latinoamericana Prensa Latina S.A. | Noticia
Noticias anteriores a enero de 2010
Suscripción AL Día
