
feb
10
Hallazgos recientes reconsideran conceptos clásicos en los complejos mitocondriales. Los avances en este campo podrían ayudar en enfermedades sin apenas tratamiento curativo.
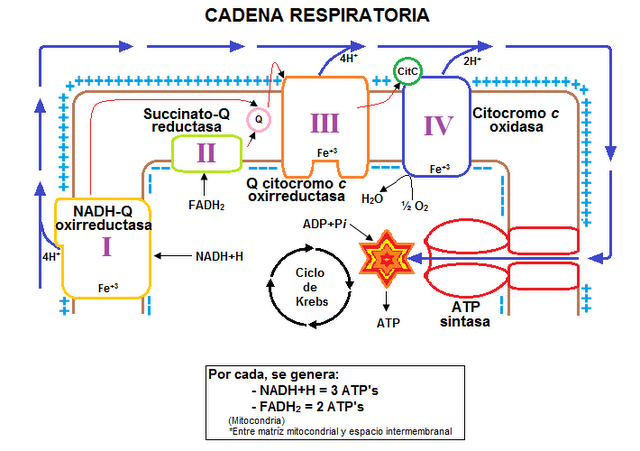 La respiración celular tiene lugar en la mitocondria mediante el sistema de fosforilación oxidativa, una maquinaria formada por cinco complejos multienzimáticos que transportan electrones, procedentes de la oxidación de sustancias como los carbohidratos y las grasas, y cuya interrelación es la responsable de generar trifosfato de adenosina (ATP), la principal fuente de energía de la célula.
La respiración celular tiene lugar en la mitocondria mediante el sistema de fosforilación oxidativa, una maquinaria formada por cinco complejos multienzimáticos que transportan electrones, procedentes de la oxidación de sustancias como los carbohidratos y las grasas, y cuya interrelación es la responsable de generar trifosfato de adenosina (ATP), la principal fuente de energía de la célula.
De un tiempo a esta parte, la comunidad científica especializada en la investigación mitocondrial asiste a un debate sobre cómo se organizan esos complejos enzimáticos -y que, por simplificar, se numeran del I al V- hasta el punto de que los nuevos hallazgos están replanteando las enseñanzas de los manuales de biología clásicos.
Una de esas científicas, Cristina Ugalde, del Instituto de Investigación Sanitaria del Hospital 12 de Octubre, en Madrid (i+12), explica que «los modelos tradicionales que sostienen que estos complejos de la cadena respiratoria funcionan libres se está sustituyendo por el concepto de que cada una de esas estructuras independientes puede asociarse formando otras mayores, los supercomplejos».
Ejercicio y mitocondria
El año pasado, dos grupos científicos, en Reino Unido y China, publicaron sendos trabajos en Nature corroborando esa idea, al describir la estructura de la cadena respiratoria, un supercomplejo conocido como respirasoma, en el que se aúna el complejo I con los complejos III y IV. Además, se está desvelando que la organización y cantidad de estos complejos mitocondriales varían dependiendo del tipo de organismo y del tejido de la célula, y ello puede influir en otros procesos celulares. Así, por ejemplo, un estudio exponía que el ejercicio físico aumenta la capacidad funcional de los respirasomas; y otro que, a mayor cantidad del complejo I en el respirasoma, más producción de radicales libres.
Una de las líneas de trabajo de Ugalde, publicada también en revistas de alto impacto, como Cell Metabolism y, más recientemente, Cell Reports, está alineada con ese revolucionario concepto. En concreto, aborda el papel de la proteína SCAFI como elemento esencial en la formación de una estructura minoritaria: la asociación de complejos III y IV. «Esto sugiere que hay proteínas específicas para la regulación fina de la formación de cada una de esas estructuras moleculares: la producción de energía y el funcionamiento de todos los tejidos del organismo dependen de ello». Y aquí cobra todo el sentido que la labor de Ugalde se realice en un hospital.
Unidades de referencia
La científica es una de las investigadoras principales del Laboratorio de Enfermedades Raras, Mitocondriales y Neuromusculares, que coordina Miguel Ángel Martín dentro del i+12. Este Instituto de Investigación, a su vez dirigido por Joaquín Arenas, incluye otras siete áreas de trabajo, todas ellas con vocación de aunar investigación clínica y básica.
Por ello, en el grupo de las enfermedades raras también se integran médicos de diferentes unidades del hospital, que, además, cuentan con la acreditación como centros de referencia (CSUR) para el diagnóstico y seguimiento de enfermedades mitocondriales tanto en niños como en adultos.
Las enfermedades mitocondriales, que en conjunto alcanzan una prevalencia estimada de uno por cada 5000 nacidos, pero para las que prácticamente no hay tratamiento curativo, se producen por alteraciones en el ADN mitocondrial (ADNmt) o en el nuclear. Martín recuerda que «ambos genomas expresan proteínas que se regulan entre sí; por ello resulta tan complejo determinar la fisiopatología de estas enfermedades». En común tienen, en principio, «un defecto en la producción de energía dentro de las células, lo que se traduce sobre todo en manifestaciones neurológicas o neuromusculares, precisamente los tejidos que más energía requieren». Lo cierto es que muchos de los procesos moleculares que rodean a estas centrales energéticas de la célula son todavía un misterio. «Una misma mutación puede producir diferentes expresiones clínicas, y no entendemos bien por qué», reconoce Martín.
Los hallazgos sobre la estructura y funcionamiento de complejos macromoleculares como el respirasoma impulsan un nuevo enfoque en la investigación de la disfunción mitocondrial, dice Ugalde. Y todo eso, sintetiza Joaquín Arenas, puede contribuir al diagnóstico diferencial de estas enfermedades desde la fases iniciales o incluso a desvelar nuevas dianas para desarrollar, por fin, tratamientos.
febrero 9/2017 (diariomedico.com)
Comments
Noticias anteriores a enero de 2010
Suscripción AL Día
